神經白塞病1例報告及文獻復習
2013-11-17 07:15:26郭俊猛耿云龍趙曉靜宋曉南
中風與神經疾病雜志 2013年2期
郭俊猛, 耿云龍, 趙曉靜, 宋曉南
白塞病為一種原因不明的以細小血管炎為病理基礎主要累及皮膚粘膜的慢性進行性多發性、多系統損害疾病,當累及神經系統時稱為神經白塞氏病(Neuro-Behcet’s disease,NBD),中樞神經受累比外周神經受累多見,神經受累的部位不同,其臨床表現亦不同。
現將我院收治的1例以腦卒中樣起病的神經白塞氏病報告如下,并對其進行回顧性分析,旨在進一步掌握其臨床特點,以期早期診斷、早期規范治療,從而提高治愈率或好轉率,最大限度地減少死亡率和致殘率。
1 病例報告
患者,女性,26歲,因左側肢體活動不靈伴飲水嗆咳10d就診。該患者于入院前10d洗澡后出現左側肢體活動不靈,表現為左上肢不能持物,下肢行走笨拙,伴有飲水嗆咳、言語笨拙,于當地醫院行頭部MRI及增強示腦干異常信號,按“腦梗死”治療后,言語笨拙感覺略有緩解,但肢體無力無明顯變化。既往:曾在2000年因反復口腔潰瘍,四肢紅斑不退,經紅斑活檢后診斷為白塞氏病,并于2005年因左側肢體間斷性活動不靈診斷為血管炎。入院時查體:T38.5℃,發育正常,智商正常,神清語明,雙側瞳孔等大同圓,直徑約2.5 mm,眼球活動自如,無眼震,對光反射靈敏,右側鼻唇溝略淺,咽反射消失,左側肢體肌力4級,左側腱反射略活躍,左側Babinski征及Chaddock征陽性,左側足背部痛覺過敏。共濟運動正常,無項強,克氏征陰性。輔助檢查:頭部MRI示:腦干梗死(急性期)(見圖1)。頭部MRA:右側大腦前動脈變細,血流信號減低。腰穿結果示:腦脊液免疫球蛋白IgG17.10IU/ml,腦脊液常規檢查:蛋白 0.35g/L,白細胞 8 ×106/L。檢驗結果:肝功、血脂、空腹血糖、風濕3項、抗核抗體、血沉、血常規、血生化、凝血常規均未見明顯異常。
2 討論
白塞病又被稱為白塞綜合征(Behcet’s syndrome,BS)、貝赫切特氏綜合征、口-眼-生殖器三聯征等,最早是在1937年由Behcet報告以前房積膿性虹膜睫狀體炎、復發性口腔粘膜潰瘍和外生殖器潰瘍為主的一組疾病,1941年Knapp報告了本病出現神經系統損害的病例;1944年Berlin作了白塞氏病的神經系統病變尸體解剖報告;1954年Cavara和D’Ermo將以中樞神經系統損害為主的白塞病命名為“神經白塞病”。
現在認為白塞病屬結締組織疾病,是一種原因不明的以細小血管炎為病理基礎,主要累及皮膚、粘膜的慢性進行性多發性多系統損害疾病,以反復發作緩解的口腔及外生殖器潰瘍、眼葡萄膜炎、結節性紅斑為特征的綜合征。BS可累及全身各種臟器,當累及神經系統時稱為神經白塞病(Neuro-Behcet’s disease,NBD)。該病例患者既往反復出現口腔潰瘍及四肢紅斑,且紅斑活檢后診斷為白塞病,后又出現神經系統癥狀。
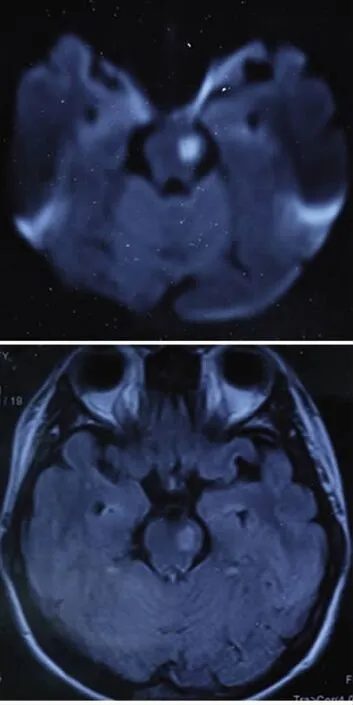
圖1 頭部MRI示DWI像為高信號,Flair像為高信號
NBD作為BD的嚴重并發癥之一,發生率約占BD患者的5% ~30%[1,2],平均為18%,最高可達 49%,男女比例約為4:1,平均患病年齡在30歲左右,發病持續時間約為4年,平均治療期是7個月,且有報道稱有5%BD患者以神經系統起病為首發癥狀。雖然對NBD的認識逐漸加深,但目前對NBD的病因及發病機制尚不明確,有學者發現有相當多的患者在發病前有一段發熱的病史,并發現在NBD患者中有扁桃體炎史的概率較正常人高,因此提出了感染學說并通過進一步研究得出 NBD可能與病毒(EB、單純胞疹、丙肝)、鏈球菌、結核桿菌有關[3]。一些學者在NBD患者的腦脊液中發現淋巴細胞增多,補體C3、IgG升高,并在血清中發現存在抗口腔粘膜細胞抗體及T4/T8之比減少和NK細胞升高而活力下降,因此提出了免疫機制學說。
NBD的發病具有明顯的地區性,以地中海、中東、東亞國家居多,通過研究發現BD的發病與人類的HLA系統有關(HLA-B51是白塞氏病的易感因素,而HLA-1類相關基因可能是決定BD發病的基因),研究發現BD與遺傳相關外,該病與環境污染、微量元素變化亦有關聯,特別是血中銅離子含量與疾病發作呈正相關。除了上述原因外,有少數學者認為性激素的分泌與鋅元素的缺乏也與本病有關。
NBD的病變形式多樣,尸體解剖和活檢標本發現其基本病理改變是顱內小血管炎,靜脈受累最重,是引起NBD病理改變的重要因素。其典型病理:大體呈腦組織水腫和腦干萎縮;在光鏡下:早期可以看見小血管周圍炎性細胞浸潤為主;晚期則表現灶性壞死,神經膠質細胞增生、局部脫髓鞘和腦膜不同程度的增生和纖維化。
NBD病變可累及中樞神經系統及周圍神經系統的各個部位,其中周圍神經受累僅占總數的1%,NBD的臨床表現中最常見的癥狀是頭痛、無力、麻木感;頭痛主要為偏頭痛,其次為緊張型頭痛。最常見的神經系統體征是反射亢進、向上跖反射、軀體感覺異常,另外錐體束征、顱神經麻痹、假性延髓征和小腦征也很常見。本例患者出現肢體肌力減弱及病理征陽性為錐體束受累所致,左側足背部出現痛覺過敏符合此病的臨床表現。臨床發現大部分NBD患者具有復發-緩解、繼發-進展、隱襲起病,原發-進展等不同發病進展形式,且同一患者不同時期可由神經系統多部位受累,故依照受累部位分為以下幾種類型:(1)腦膜腦炎型;(2)腦干型:常以腦血管意外為主要發病形式,主要是腦內血管病變所致;(3)脊髓型;(4)周圍神經病變型;(5)小腦病變型:常以小腦共濟失調為主要表現形式;(6)顱神經麻痹型:外展神經、面神經受累較多。此外還可表現良性高顱壓[4],近年還有肌肉受累的報道。其中腦干型最為常見,其腦干病變由于炎性閉塞的血管較小,損害范圍往往小于典型的由梗死所致的腦干綜合征。本病例患者屬于腦干型。
目前白塞氏綜合征的診斷依據參照1998年第八屆國際白塞氏病學術大會國際研究組制定新的白塞氏病診斷標準,而NBD目前沒有獨立的診斷標準,臨床上一般將神經系統的表現和白塞氏病的診斷標準結合起來診斷。實驗室檢查中CSF對NBD的診斷有重要參考價值,文獻報道部分NBD患者腦脊液淋巴細胞增多,但總數小于60個/dl,蛋白量也高于正常,但均低于100mg/dl[5],此外還會在部分NBD患者的腦脊液中發現髓鞘堿性蛋白的增多。有時在缺乏中樞神經系統受累癥狀的BD患者的腦脊液中發現白細胞、淋巴細胞和蛋白增高,這提示有亞臨床型的NBD存在。在其它實驗室檢查指標中,患者血清及腦脊液中的AMSF、ESR的增高均可提示NBD的活動性,并將NBD患者血清和腦脊液中的IL-6作為監測治療的指標。
目前MRI是診斷神經白塞氏病腦損害最敏感的方法了,MRI的改變反映了白塞氏病的組織學變化。急性期是一個急性炎癥過程,病灶T1加權低信號、T2加權高信號,常常位于腦橋、中腦、小腦、基底節區、腦室周圍白質(通常不靠近腦室壁),其中錐體束最常受累,尤其是腦橋及中腦的錐體束。高信號病灶可能反映了急性炎癥的過程,并有可能因為強化及中位效應而極像腫瘤,這可能是炎癥破壞血腦屏障和血管源性水腫導致的,這些高信號病灶在經過有效治療后會變小或消失。所以NBD是一種中樞神經系統的血管性炎癥的疾病,其MRI的表現有“可逆性”的特點,急性期過后或經治療后病灶體積可縮小或消失,“錐體束征”是其重要特點,特別是腦橋和中腦的皮質脊髓束,這是因為NBD累及的靜脈都是腦干軸向靜脈所致。在慢性期或者該病后期MRI可發現顱后窩結構萎縮而大腦半球不萎縮的表現,且病灶信號強度減弱,這可能與含鐵血黃素沉積有關。該患者出院后將繼續隨訪,復查MRI可證實其“可逆性”影像學改變。
目前仍未找到根治神經白塞氏病的治療方法,現階段主要是用激素、秋水仙素、細胞毒素類藥物(如環磷酰胺、氨甲喋呤、干擾素和氨苯礬)和血漿置換,但主要是對BD患者有效,對NBD患者效果欠佳。從臨床類型看,脊髓型和周圍神經病變型預后相對較好,腦膜腦炎型和腦干型預后較差,早期診斷及治療有可能改善預后。
[1]Borhani Haghighi A,Pouimmand R,Nikseresht AR.Neuro-Behcet disease[J].A review Neurologist,2005,11(2):80-89.
[2]Ho CL,Deruytter MJ.Manfestations of Neuro-Behect’s disease.Report of two cases and review of the literature[J].Clin Neurol Neurosurg,2005,107(4):310-314.
[3]徐 雁綜述.神經白塞氏病[J].國外醫學神經病學神經外科學分冊,1998,25(3):157.
[4]Siva A,Altintas A,Saip S.Behcet’s syndrome and the nervous system[J].Current Opinion Neurolog,2004,17(3):347-357.
[5]Ohta K,Obara K,SatoH,et al.Diffusion weighted magnetic resonance imaging of Neuro Behcet’s disease[J].Rinsho Shinkeigaku,2000,40(4):398-401.
