1, 8-二氮雜雙環(5, 4, 0)-7-十一烯的合成工藝及其應用開發
2013-07-18 07:09:54馮筱晴王瑞瑞王彥臣宋國強
化工進展 2013年1期
馮筱晴,沈 力,王瑞瑞,王彥臣,宋國強
(1常州大學制藥與生命科學學院,江蘇 常州 213164;2常州市藥品制造與質量控制工程重點實驗室,江蘇 常州213164)
光聚合技術是 20世紀 60年代發展起來具有“5E”特點:高效(efficient)、適應性廣(enabling)、經濟(economical)、節能(energy saving)和環境友好(environmental friendly)的一種新型材料表面加工技術。光產堿劑(PBG)是繼自由基光引發劑和光產酸劑之后發展出的一種光引發劑,屬于光固化體系中的關鍵組分,基于其良好的抗氧阻性和抗基板腐蝕性,在微電子行業和高端表漆中得到廣泛的應用[1-4]。新型取代芐基脒類光產堿劑以1, 8-二氮雜雙環(5, 4, 0)-7-十一烯(DBU)這種雙脒環類化合物為結構主體,光解后釋放強堿DBU,引發效率高可催化含有環氧樹脂或脂肪族異氰酸酯類配方,由此可期待此類PBG擴展光聚合配方的應用前景,實現DBU的應用價值提升[5-7]。
目前文獻報道的 DBU的合成方法主要有氮丙啶-內酰胺法、內酰胺-丙烯腈霍夫曼反應法、內酯-烯化二胺法等,相對于內酰胺-丙烯腈加氫環合法,前3種方法原料價格較高,反應條件苛刻,總收率較低且研究報道較少[8-12]。本工作重點研究內酰胺-丙烯腈加氫環合制備工藝,通過對反應溫度、催化劑用量、投料比等影響因素的考察,改進工藝以較高收率和純度得到DBU。此工藝分為3個步驟:①由ε-己內酰胺和丙烯腈合成N-(β-氰乙基)-ε-己內酰胺(CEC,Ⅰa);②將CEC催化加氫合成N-(γ-氨基丙基)-ε-己內酰胺(APC,Ⅰb);③APC脫水環化、減壓蒸餾,即可得DBU(Ⅰc)[12-13]。DBU的合成路線見圖1。
再以DBU(Ⅰc)為原料,分別和5種取代基氯化芐經過先取代成氯鹽再還原的方法,合成5種取代芐基脒類光產堿劑Ⅱa、Ⅱb、Ⅱc、Ⅱd和Ⅱe。分別考察不同溶劑,不同溫度及原料投料比對兩步反應的影響。研究表明:此合成工藝,第一步成鹽收率高,第二步采用氫化鋰鋁為還原劑還原活性強,操作簡單,可高純度高收率地得到5種取代芐基脒類光產堿劑,為此系列PBG的合成作為參考[14-15]。PBGs的合成路線見圖2。

圖1 DBU的合成路線
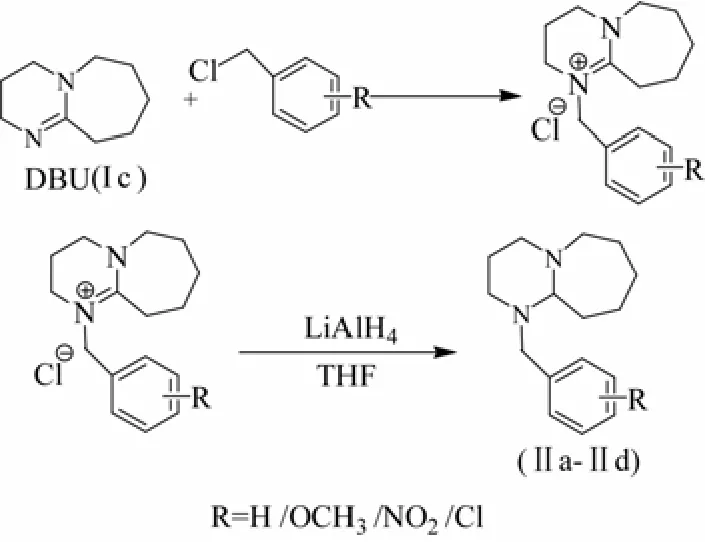
圖2 取代芐基DBU類光產堿劑的合成路線
1 實驗部分
1.1 儀器和試劑
Bruker DRX 500MHz核磁共振儀(德國Bruker公司),溶劑為DCCl3;GC-2010型氣相色譜儀(日本島津);Agilent 1260型液相色譜儀(美國安捷倫公司)。
ε-己內酰胺、丙烯腈、對苯二酚、對甲苯磺酸、二甲苯、氯芐、4-氯代氯芐、4-硝基氯芐、3-甲氧基氯芐、4-甲氧基氯芐等均為分析純級試劑,鎳、甲苯、THF、二氯甲烷、甲醇、氫氧化鉀、氫化鋰鋁、無水硫酸鎂等均為工業級,含量均≥99.0%。
1.2 產物分析色譜條件
液相色譜:Shim-pack CLC-ODS 6.0 mm× 150 mm、1.0 mL/min流速、柱溫25 ℃、流動相90%甲醇/水、檢測波長254 nm。
氣相色譜:AC-1、FID1檢測器,溫度250 ℃、SPL1溫度280 ℃、柱箱溫度210 ℃、總流量51.5 mL/min、柱流量1.21 mL/min、壓力150.0 kPa、分流比39.0、氮氣載氣。
1.3 DBU(Ⅰc)的合成
1.3.1N-(β-氰乙基)-ε-己內酰胺(Ⅰa)的合成
向250 mL帶機械攪拌的反應瓶中投入22.6 g(0.20 mol)ε-己內酰胺,120 mL甲苯為溶劑,再加入0.56 g(0.01 mol)氫氧化鉀。在恒壓漏斗中放入16.9 g(0.24 mol)丙烯腈和少量對苯二酚,待ε-己內酰胺融化時滴加丙烯腈,20 min內滴完,油浴控制反應溫度在70 ℃左右,保溫反應24 h,停止反應,有機相水洗至中性,無水MgSO4干燥,過濾,旋蒸除去溶劑,減壓蒸餾得產品34.0 g,收率98.1%,純度97.8%。
1.3.2N-(γ-氨基丙基)-ε-己內酰胺(Ⅰb)的合成
取 30 g(0.18 mol)N-(β-氰乙基)-ε-己內酰胺加入高壓釜中,250 mL無水甲醇為溶劑,再加入30 g鎳,通入氫氣,保持釜內壓力2 MPa,攪拌機轉速450 r/min,保持溫度80~100 ℃反應5 h。后處理:將反應料液抽濾除去鎳,干燥,旋蒸得到產品27.9 g,收率91.1%,純度98.0%。
1.3.3 APC成環反應
向250 mL帶機械攪拌的反應瓶中投入20.0 g(0.12 mol)N-(γ-氨基丙基)-ε-己內酰胺(APC)和1.03 g對甲基苯磺酸,120 mL二甲苯為溶劑,油浴控溫150~160 ℃反應8 h,停止反應,向燒瓶中加入少量水停止反應,有機相水洗至中性,無水MgSO4干燥,過濾,減壓旋蒸除去溶劑得產品17.4 g,收率91.4%,純度大于95.4%。
Ⅰc的1HNMR(CDCl3,500MHz):δ為3.295~3.273(2H, t,—CH2);3.231~3.186(4H, m,—CH2);2.417~2.397(2H, t, —CH2);1.825~1.778(2H, m, —CH2);1.670~1.661(4H, m, —CH2);1.655~1.576(2H, m, —CH2),LC-MS MS(m/z)為 152[M]+。
1.4 8-芐基-1, 8-二氮雜雙環[5.4.0]十一烷(Ⅱa)的合成
1.4.1 取代合成氯鹽反應
在帶攪拌的 100 mL四口燒瓶中,稱取 4.0 g DBU,加入50 mL甲苯,室溫下攪拌。取3.5 g氯芐,以20 mL甲苯溶解,轉移至25 mL的恒壓滴液漏斗內,約30 min滴完,升溫至 90 ℃,反應6 h,抽濾,濾餅用新鮮甲苯洗滌,除去溶劑得白色固體季胺鹽7.2 g。
1.4.2 亞胺還原反應
在帶攪拌的100 mL四口燒瓶中,取4.0 g白色固體季胺鹽,加入50 mL無水THF。控溫25 ℃攪拌反應,稱取0.273 g氫化鋰鋁分批加至燒瓶中,攪拌過夜,薄層層析(TLC)磷鉬酸顯色跟蹤反應至原料反應完全。后處理:料液溫度降至0 ℃,依次滴加1.0 mL水,1 mL 10%的NaOH水溶液,1.0 mL水,繼續反應1 h。反應料液抽濾,收集有機相,蒸除溶劑,剩余物用乙酸乙酯溶解,少量去離子水洗2~3遍,收集有機相干燥,過濾,旋蒸得淺黃色油狀液體 2.7 g產品,產率 76.7%,液相分析純度93.2%。
Ⅱa的1H NMR(CDCl3,500MHz):δ為7.365~7.353(1H, d,—C6H5);7.301~7.240(2H, q,—C6H5);7.234~7.221(2H, d, —C6H5);3.828~3.821(2H, d, —CH2);3.107~3.041(1H, m, —N—CH);2.981~2.831(4H, m, —CH2);2.423~2.408(3H, q, —CH2);2.254~2.217(4H, m, —CH2);2.047~1.463(5H, m, —CH2),LC-MS MS(m/z)為 245[M+1]+。
1.5 8-(4-氯芐基)-1, 8-二氮雜雙環[5.4.0]十一烷(Ⅱb)的合成
以4-氯代氯化芐和DBU為原料按照1.4節中的方法合成產品Ⅱb,Ⅱb的1HNMR(CDCl3,500MHz),δ為 7.289~7.249(4H, m, —C6H4);3.853~3.824(2H, d, —N—CH2);3.487~3.345(1H, m, —CH2);3.031~2.924(2H, m, —CH2);2.776~2.642(2H, m, —CH2);2.563~2.518(2H,m, —CH2);2.418~1.236(10H, m, —CH2),LC-MS MS(m/z)為 277[M+1]+。
1.6 8-(4-甲氧芐基)-1, 8-二氮雜雙環[5.4.0]十一烷(Ⅱc)的合成
以4-甲氧氯化芐和DBU為原料按照1.4節中的方法合成Ⅱc,Ⅱc的1HNMR(CDCl3,500 MHz),δ為 7.262~7.238(2H, t, —C6H4);6.851~6.834(2H, q, —C6H4);3.833~3.792(2H, q, —N—CH2);3.432~3.345(2H, m, —OCH2);3.036~2.921(2H,m, —CH2);2.809~2.784(2H, m, —CH2);2.689~2.635(2H, m, —CH2);2.388~1.391(10H, m,—CH2),LC-MS MS(m/z)為 275[M+1]+。
1.7 8-(3-甲氧芐基)-1, 8-二氮雜雙環[5.4.0]十一烷(Ⅱd)的合成
以3-甲氧氯化芐和DBU為原料按照1.4節中的方法合成Ⅱd,Ⅱd的1HNMR(CDCl3,500 MHz),δ為 7.278~7.232(1H, t, —C6H4);7.221~7.1-2(1H, t, —C6H4);6.841~6.824(2H, q,—C6H4);3.801~3.792(2H, q,—N—CH2);3.422~3.335(2H,m, —OCH2);3.028~2.897(2H, m,—CH2);2.796~2.763(2H, m,—CH2);2.673~2.583(2H, m,—CH2);2.368~1.372(10H, m,—CH2),LC-MS MS(m/z)為 275[M+1]+。
1.8 8-(4-硝基芐基)-1, 8-二氮雜雙環[5.4.0]十一烷(Ⅱe)的合成
以4-硝基氯化芐和DBU為原料按照1.4中的方法合成Ⅱe,Ⅱe的1HNMR(CDCl3,500MHz),δ為 8.133~7.262(4H, m,—C6H4);3.975~3.943(2H, d,—CH2);3.238~3.207(2H, d,—CH2);3.118~3.102(2H, q,—N—CH);2.752~1.406(14H,m,—CH2), LC-MS MS(m/z)為 290[M+1]+。
2 結果與討論
2.1 N-(β-氰乙基)-ε-己內酰胺(Ⅰa)的合成
(1)反應溫度的選擇 通常情況下較高反應溫度有利于ε-己內酰胺和丙烯腈反應,而溫度過高丙烯腈容易發生自聚,所以反應中加入少量對苯二酚防止發生聚合[12]。考察以氫氧化鉀為催化劑,不同反應溫度的影響,實驗結果見表1。
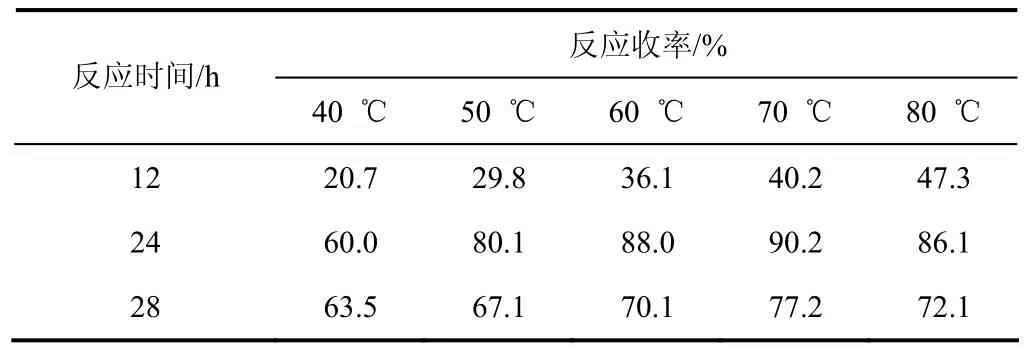
表1 溫度對反應結果的影響
由表1可以看出:反應溫度小于70℃時,隨著反應溫度的提高,縮合反應收率逐漸提高;當反應溫度高于70 ℃,反應24 h后,雖然原料轉化率依然較高,但已產生部分聚合產物,目標產物收率降到了 86.1%,同樣反應時間增長也使得聚合產物迅速增多。綜合考慮,溫度應控制在65~70 ℃,反應24 h較佳。
(2)投料比的選擇 通過改變投料比,試圖提高縮合反應的收率,由于丙烯腈的價格較便宜,在70℃條件下,通過改變丙烯腈的量來考察反應,實驗結果見表2。

表2 投料比對反應結果的影響
由表2可以看出:70 ℃條件下,隨著丙烯腈的量增加,縮合反應收率逐漸提高;當ε-己內酰胺和丙烯腈的摩爾投料比達到1∶1.2時,Ⅰa的反應收率達到最高,繼續增加丙烯腈的量,收率變化幅度不明顯。根據原料經濟學原理,最終確定反應投料比為1∶1.2。
2.2 N-(γ-氨基丙基)-ε-己內酰胺(Ⅰb)合成
在高壓釜中以鎳加氫法對 Ⅰa進行還原,從而合成N-(γ-氨基丙基)-ε-己內酰胺(Ⅰb),控制氫氣量一定情況下,考察不同溫度的影響,實驗結果見表3。
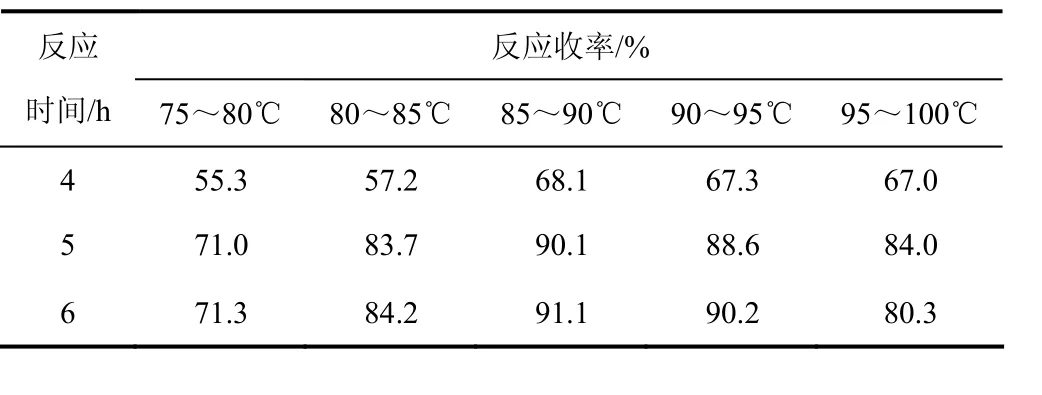
表3 溫度對反應結果的影響
由表3可以看出:在反應溫度小于90 ℃時,隨著反應溫度的提高,有利于反應,還原產物Ⅰb收率逐漸提高;當溫度高于90 ℃后,反應5 h,氣相分析副產物增多,Ⅰb收率反而下降,而當反應溫度控制在85~90 ℃,反應6 h,反應收率變化較小。綜合考慮,控制溫度在85~90 ℃反應5 h較佳。
2.3 APC成環反應
(1)反應時間的選擇 以對甲基苯磺酸為催化劑,二甲苯為溶劑,在高溫下使N-(γ-氨基丙基)-ε-己內酰胺(Ⅰb)成環生成DBU,氣相監測,考察反應時間的影響,實驗結果見表4。

表4 反應時間對反應結果的影響
由表4可以看出:隨著反應時間變長,DBU(Ⅰc)的收率逐漸提高,7~8 h后反應達到平衡,綜合考慮反應轉化率及能耗問題,確定反應時間為7.5 h。
(2)催化劑量的選擇 在150 ℃反應7.5 h的條件下,考察催化劑用量對反應的影響,實驗結果見表5。
由表5可以看出:在其它條件不變的情況下,增加對甲基苯磺酸的量,可以提高 DBU的收率,當其用量達到1.6 g時,反應收率最高達到97.3%,繼續增加時,反應趨于平衡,Ⅰc收率增加幅度不明顯,考慮到催化劑用量過多,原料和酸性廢水后處理成本增加,最終確定催化劑用量為1.6 g,相當于反應底物量的8%。

表5 催化劑量對反應結果的影響
2.4 8-芐基-1, 8-二氮雜雙環[5.4.0]十一烷(Ⅱa)的合成
(1)取代成鹽反應溫度的選擇 在甲苯中以等摩爾量的DBU(Ⅰc)和氯芐反應生成8-芐基-1,8-二氮雜雙環[5.4.0]-7-十一烯氯鹽,通常情況下較高反應溫度有利于氯鹽生成。考察反應溫度的影響,實驗結果見表6。
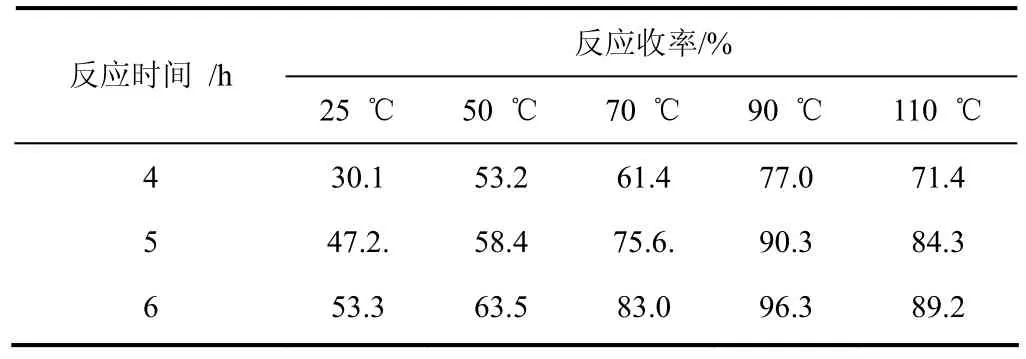
表6 溫度對反應結果的影響
由表6可以看出:隨著反應溫度的升高,氯鹽收率明顯提高,當溫度達到90 ℃反應6 h,收率最高達到 96.3%;繼續升高溫度直至甲苯回流,液相跟蹤顯示氯芐已經反應完全,然而氯鹽收率反而下降,由此說明氯芐在較高溫度環境中不穩定,容易分解,不參與氯鹽生成,因此綜合考慮,反應溫度應控制在90~95 ℃反應6 h較佳。
(2)取代成鹽反應投料比的選擇 在90 ℃甲苯溶劑中,通過改變投料比,提高成鹽反應的收率,實驗結果見表7。由表7可以看出:90 ℃條件下,隨著 DBU或氯芐的量增加,成鹽反應收率都相應提高,當摩爾投料比達到1∶1.05時,氯鹽的反應收率達到 98.7%,繼續增加氯芐的量,氯鹽收率變化已經不明顯,相對于提高 DBU量得到的反應效果較好,原料經濟。因此,最終確定反應投料比為1∶1.05。

表7 投料比對反應結果的影響
(3)亞胺還原反應投料比的選擇 以氫化鋁鋰為還原劑在THF中,室溫下將8-芐基-1, 8-二氮雜雙環[5.4.0]-7-十一烯氯鹽中的亞胺雙鍵還原,考察原料投料比對反應收率的影響,實驗結果見表8。
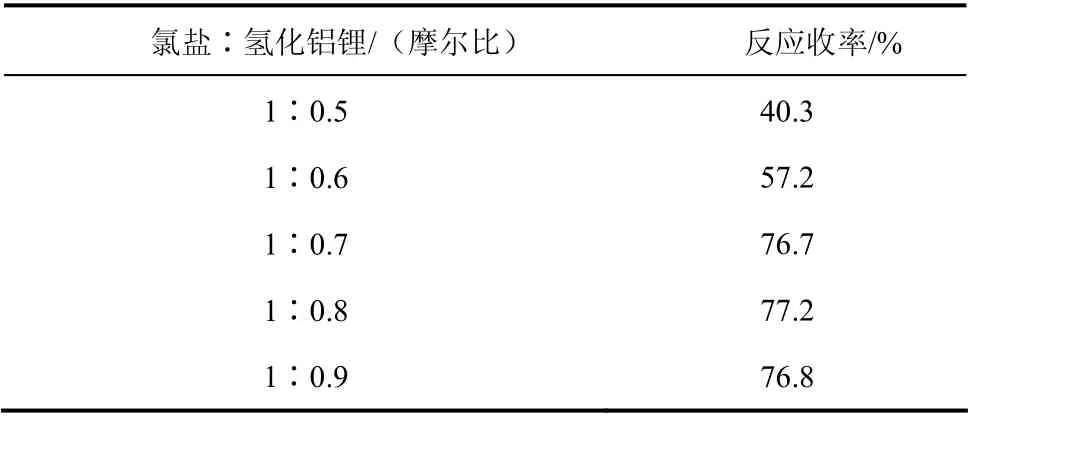
表8 投料比對反應結果的影響
由表8可以看出:氫化鋁鋰還原性強,室溫條件下,隨著還原劑量的增加,亞胺還原反應收率相應提高;當反應摩爾投料比達到1∶0.7時,Ⅱa的反應收率達到 76.7%,繼續增加還原劑的量,收率變化已經不明顯。因此從原料經濟學考慮,最終確定氯鹽和氫化鋁鋰的反應投料比為1∶0.7。
2.5 其它幾種取代芐基脒類光產堿劑的合成工藝優化結果
以合成出的DBU(Ⅰc)和4種帶不同取代基團的氯化芐為原料,經先取代成鹽再還原的方法,成功合成其它4種取代芐基DBU類光產堿劑,通過對溶劑,成鹽溫度,投料比及其它反應條件的考察,得到這4種PBGs的工藝優化結果見表9。
綜上得出結論:當在氯化芐苯環上取代硝基時可以明顯增強芐基碳正離子活性,使其與DBU 反應成鹽活性增加,反應溫度降低,時間縮短,產率較高;而當含有給電子基時,芐基活性相應降低,投料比需增加。
3 結 論
(1)通過單因素試驗法,得到了目標產物Ⅰa,Ⅰb和DBU(Ⅰc)的較佳合成工藝條件為:以ε-己內酰胺和丙烯腈在甲苯中合成Ⅰa,溫度對此縮合反應的影響較大,ε-己內酰胺和丙烯腈按1∶1.2投料,70 ℃條件下反應24 h,Ⅰa收率98.1%,純度大于97.8%;以Ⅰa為原料在85~90℃經鈀碳加氫還原5 h得Ⅰb,收率91.1%,純度大于98.0%;以Ⅰb量8%的對甲苯磺酸催化,反應7.5 h得DBU(Ⅰc),收率97.3%,純度大于95%,此工藝得到改進。

表9 其它4種PBGs的合成工藝優化結果
(2)以DBU(Ⅰc)和氯化芐為原料(摩爾比為1∶1.1)甲苯為溶劑在90 ℃下反應6 h,氯鹽收率近100%,以0.7當量的氫化鋁鋰常溫下還原氯鹽得Ⅱa,收率76.7%,純度93.2%。
(3)以DBU(Ⅰc)分別和甲氧基、氯基、硝基取代的氯化芐為原料,選用不同溶劑、投料比、溫度及還原劑量合成Ⅱb、Ⅱc、Ⅱd、Ⅱe,收率均達到70%以上,提高了DBU的產品附加值。結果表明:經硝基取代的氯芐反應活性最高,成鹽反應溫度低;以 DBU(Ⅰc)和取代基氯芐經先成鹽再還原的方法,可成功合成此類取代芐基 DBU類光產堿劑。此方法原料來源廣泛,操作簡便,收率相對較高,純度較好,可為此類化合物的工業化生產奠定理論基礎。
[1]姚梅,王汝敏,董萌.紫外光固化體系的研究進展[J].中國膠黏劑,2006,15(6):33-36.
[2]魏杰金,養智.光固化涂料[M].北京:化學工業出版社,2005.
[3]余宗萍,廖申偉,羅榮榮.常用光引發劑在紫外光固化涂料中的應用研究[J].上海涂料,2010,48(8):13-16.
[4]Shi Suqing,Gao Hong,Wu Gangqiang,et al.Cyclic acetal as coinitiator for bimolecular photoinitiating systems[J].Polymer,2007,23(48):2860-2865.
[5]Baudin G,Dietliker K,Jung T.Photoactivable nitrogen bases:US,7538104B2[P].2009.
[6]Edward J Urankar,Jean M J Frechet.Photogenerated base in polymer curing and imaging:Cross-linking of base sensitive polymers containing enolizable pendant groups[J].Chem.Mater., 1997,9(12):2861-2868.
[7]Kutal C,Willson C G.Photoinitiated cross-linking and image formation in thin polymer films containing a transition metal compound[J].The Journal of the Electrochemical Society,1987,134:2280.
[8]Naoya Kumagai,Shigeki Matsunaga.An efficient synthesis of bicyclic amidines by intramolecular cyclization [J].Angewandte Chemie International Edition,2004,43(4):478-482.
[9]Noboru Matsumura,Hiroshi Nishiguchi,Masao Okada,et al.Preparation and characterization of 6-substituted 1,8-diazabi-cyclo[5.4.0]undec-7-ene[J].Journal of Heterocyclic Chemistry,1986,23(3):885–887.
[10]Armin Guggisberg,Urs Kramer, Christian Heidelberger.Umamidierungsreaktionen an cyclischen amino-amid-systemen.3 Mitteilung über umamidierungsreaktionen [J].Helvetica Chimica Acta,1978,61(3):1050–1063.
[11]馬震,馬斌,楊文庫.1,8-二氮雜雙環(5,4,0)-7-十一烯的制備方法:中國,200310115849.5[P].2003.
[12]趙會吉,邢金仙,殷長龍.辛二腈加氫制取辛二胺的研究[J].石油大學學報,2001,25(3):59-61.
[13]陽海,龐懷林,廖文文.催化加氫合成6-氨基-7-氟-2H-1,4-苯并惡嗪-3(4H)-酮[J].精細化工,2006,23(11):1142- -1144.
[14]鐘榮,曾兆華,楊建文,等.光產堿反應及其應用[J].高分子通報,2003(2):43-49.
[15]許文杰,楊建文,曾兆華,等.光敏性季銨鹽的合成與性能研究[J].功能高分子學報,2002,15(2):128-130.
