石油焦基活性炭的制備及甲烷吸附性能
2013-03-03 05:52:18郭紅娜張永春
化工進展 2013年1期
關(guān)鍵詞:改性
郭紅娜,張永春
(大連理工大學(xué)化工學(xué)院,遼寧大連116024)
我國煤層氣資源儲量豐富,居世界第三位[1],但其利用率低,主要原因是低濃度甲烷的提純分離問題沒有解決[2-3]。在煤礦開采過程中,有70%的煤層氣通過煤礦乏風(fēng)排出[4]。甲烷的溫室效應(yīng)是二氧化碳的2123倍,對臭氧的破壞能力是二氧化碳的7倍;同時高濃度的煤層氣不僅可以作為民用及工業(yè)原料,還可以當(dāng)做化工原料用來生產(chǎn)C1化工產(chǎn)品及合成油[5]。因此,研究低濃度煤層氣的濃縮與分離引起了人們越來越多的關(guān)注。
目前,常用的煤層氣濃縮與分離的方法主要有低溫蒸餾法[6]、吸附分離法及膜分離法[7]等。其中吸附分離法具有能耗低、流程簡單、操作簡便、環(huán)境效益好等優(yōu)點[8]。吸附分離法的關(guān)鍵是吸附劑的選擇,對吸附劑的要求是大的比表面積、高的吸附容量、良好的選擇吸附能力、較高的機械強度及再生性。活性炭以其便宜的價格、良好的分離效果,受到了人們廣泛的關(guān)注。王琰[9]制備的TJU01活性炭,在296 K和0.4 MPa下對甲烷的吸附量為0.57 mmol/g。張曉環(huán)[10]用磷酸活化椰殼炭化料制備的活性炭,在283 K和0.1 MPa下對甲烷的吸附量是7.149 m L/g。雷利春[4]選用商業(yè)AC-1在298 K和0.5 MPa下對甲烷的吸附量為12.925 m L/g。Yuan等[11]用軟模板法制備了一種中孔碳,在298 K、0.1 MPa下甲烷的吸附量為1.05 mmol/g。Liu等[12]研究發(fā)現(xiàn),微孔范圍在0.71.3 nm的活性炭更利于甲烷的分離。
本文以盤錦石油焦為原料制備的高比表面積活性炭(HSAAC)為吸附劑,采用動態(tài)吸附實驗裝置,考察了不同預(yù)處理條件及活化方法對甲烷吸附性能的影響,并研究了粉末活性炭成型情況及再生性能,為分離濃縮低濃度甲烷技術(shù)提供應(yīng)用基礎(chǔ)。
1 實驗部分
1.1 實驗試劑
石油焦(PC),工業(yè)品;KOH、羧甲基纖維素鈉 (CMC)、高氯酸、雙氧水、鹽酸、硝酸等均為分析純;石油焦基活性炭,自制;N2純度為99.999%,CH4/N2混合氣中甲烷濃度為0.993%。
1.2 吸附劑的制備及表征
1.2.1 石油焦的預(yù)處理
配制20%的氧化劑溶液,將氧化劑與PC按照一定液固比,在一定的改性溫度下改性一定時間,后水洗至中性、110℃烘箱中干燥3 h,得到預(yù)處理PC。經(jīng)H2O2改性后的石油焦記為H2O2-PC。
1.2.2 活性炭的制備
KOH與PC按一定質(zhì)量比混合均勻,在馬弗爐中N2氣氛保護下800℃活化1 h,冷卻至室溫,所得固體先酸洗后水洗至濾液為中性,110℃烘箱中干燥3 h得石油焦基活性炭。未經(jīng)處理的石油焦制備的活性炭記為AC,H2O2處理的石油焦制備的活性炭記為H2O2-AC。
1.2.3 活性炭的成型
將CMC、水和活性炭粉末按一定的質(zhì)量比混合均勻,置于帶孔磨具中,用壓片機在一定壓力下擠成直徑為2 mm的條,經(jīng)60℃干燥3 h,200℃焙燒1 h得柱狀活性炭,記為CMC-AC。
1.2.4 石油焦與活性炭的表征
樣品的比表面積、孔容及孔徑分布利用北京精微高博科技有限公司JW-BK型靜態(tài)吸附比表面測試儀測定,采用BET公式計算比表面積,HK公式計算微孔孔徑分布及孔容。用Thermo公司Nicolet Nexus型傅里葉變換紅外光譜儀分析樣品的結(jié)構(gòu)。利用大連化工研究設(shè)計院的DLⅡ型智能顆粒強度測定儀測定條狀活性炭的軸向強度和徑向強度。
1.3 甲烷吸附裝置及穿透曲線測定
實驗采用動態(tài)法吸附實驗裝置 (圖1)測定甲烷的穿透曲線。

圖1 動態(tài)吸附實驗裝置
首先通入氮氣對吸附柱沖壓至0.6 MPa,關(guān)閉氮氣;使原料氣以100 m L/min的流速通過裝有2 g吸附劑的吸附柱;出口氣體濃度用福立GC-9790(FID檢測器)在線分析,得到不同吸附時間下流出氣體的組成,以出口氣體組成對應(yīng)吸附時間作圖得甲烷的穿透曲線。甲烷穿透吸附量和飽和吸附量根據(jù)吸附穿透曲線計算得到[13]。
2 結(jié)果與討論
2.1 預(yù)處理石油焦制備活性炭的正交實驗結(jié)果及分析
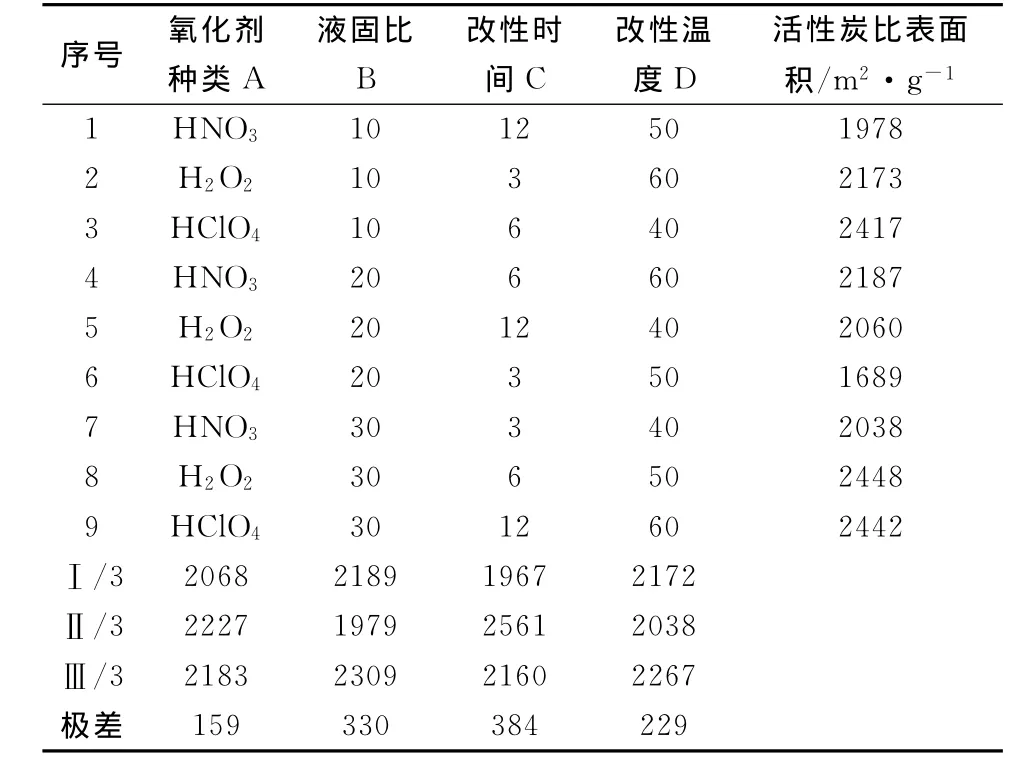
表1 預(yù)處理石油焦制備活性炭的正交實驗設(shè)計及結(jié)果
從表1可以看出,極差大小順序為C>B>D>A,即在4個因素中,對活性炭比表面積影響的大小順序為:改性時間>液固比>改性溫度>氧化劑種類;對設(shè)計的三水平而言,最利于HSAAC制備的工藝參數(shù)組合是A2B3C2D3,即以H2O2為氧化劑、液固比為30 m L/g,改性時間為6 h,改性溫度為60℃。實驗在堿焦比為3的條件下所得活性炭的最大比表面積是2448 m2/g。
2.2 雙氧水改性石油焦方法不同對活性炭吸附性能的影響
2.2.1 雙氧水改性時間的影響
在石油焦制備HSAAC的預(yù)處理階段加入氧化劑可以改變其表面官能團和酸堿性,官能團的改變會對活化過程造成影響,從而改變活性炭的吸附性能。圖2為雙氧水作氧化劑,堿焦比為3時,改性時間不同對甲烷吸附性能的影響。從圖2中可以看出,雙氧水預(yù)改性石油焦可以提高活性炭的甲烷吸附性能,說明了雙氧水作氧化劑,對石油焦表面官能團具有一定的修飾作用;并且穿透時間隨改性時間的延長而增加,當(dāng)預(yù)處理時間大于6 h后,增長的趨勢變得緩慢,考慮到其經(jīng)濟性和可行性,預(yù)處理時間為6 h較適合。
圖3是雙氧水改性前后石油焦的紅外譜圖。3450 cm-1處譜峰是O—H伸縮振動,歸屬為樣品表面羥基或者吸附的水,1638 cm-1處譜峰是碳材料的特征峰,歸屬于苯環(huán) ═C C鍵的伸縮振動,1398 cm-1處峰歸屬為烷基基團變形振動,1200 cm-1左右是C—O—C、C—O的伸縮振動。與未經(jīng)改性的石油焦相比,經(jīng)過雙氧水處理的石油焦在波數(shù)1398 cm-1處的峰強度減弱,說明經(jīng)過雙氧水預(yù)處理后,石油焦的表面烷基基團減少。Jiang等[17]認為,在改性過程中,石油焦表面的烷基基團被氧化成含氧官能團。含氧官能團的增加利于石油焦的活化,從而提高了活性炭的性能。
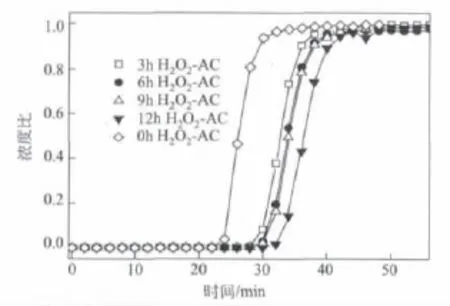
圖2 改性時間不同對甲烷吸附性能的影響

圖3 H 2 O2處理前后石油焦的紅外譜圖
2.2.2 液固比的影響
圖4是不同液固比下,堿焦比為3時預(yù)處理石油焦制備活性炭的甲烷吸附穿透曲線。從圖4可以看出,用雙氧水處理過的石油焦,活性炭的甲烷吸附量有明顯提高。但是液固比為10 m L/g與20 m L/g時,甲烷的吸附量無明顯差異;當(dāng)液固比增至30 m L/g時,甲烷的吸附量有所下降。綜合考慮,液固比為10 m L/g時較適合。
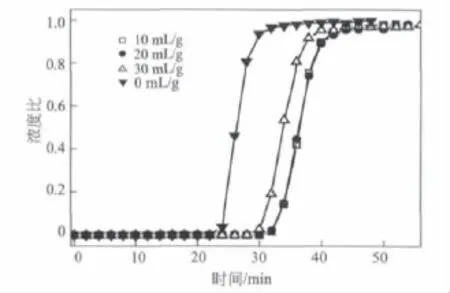
圖4 液固比對甲烷吸附性能的影響
2.3 石油焦活化過程對活性炭甲烷吸附性能的影響
2.3.1 石油焦粒度的影響
圖5是堿焦比為4時不同石油焦粒度下制備活性炭的甲烷吸附穿透曲線圖。從圖5可以看出,石油焦粒度為6080目時,其甲烷的吸附量最高;當(dāng)石油焦粒度小于80目時,粒度的改變對制得活性炭的甲烷吸附量影響不大。
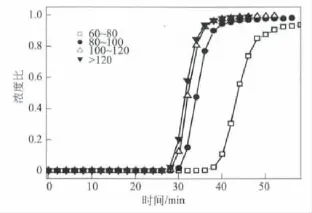
圖5 石油焦粒度不同對甲烷吸附性能的影響
表2是堿焦比為4時不同粒度石油焦制備活性炭的比表面積、孔容積及孔徑大小。從表2可以看出,活性炭的比表面積在2230 m2/g以上,微孔容積占總孔容積的比例在80%左右;隨著原料粒度的減小,活性炭的比表面積、孔容及孔徑有上升趨勢。當(dāng)原料粒度過細時,原料與活化劑KOH的混合更均勻,活化時活化程度加深,活性炭的比表面積變大、孔結(jié)構(gòu)更加發(fā)達,孔徑變大。孔徑變大對甲烷的吸附力變?nèi)酰辜淄榈奈搅拷档停f明只有合適的孔徑才利于甲烷的吸附,這與Liu等[12]的研究一致。

表2 不同粒度石油焦制備活性炭的比表面積、孔容積及孔徑
2.3.2 活化載氣流速的影響
圖6是堿焦比為4時不同活化載氣流速下制備活性炭的比表面積及甲烷的吸附量圖。從圖6可以看出,載氣流速為100 m L/min時,甲烷的穿透吸附量為13.81 m L/g。當(dāng)流速增大時,甲烷的吸附量有所下降,同時,制得活性炭的比表面積有下降的趨勢,這是因為活化時氣體流速增大,活化載氣可以降低反應(yīng)過程生成氣體的濃度,使活化程度加深,造成活性炭比表面積的降低。當(dāng)氣體流速為300 m L/min時,活性炭的比表面積進一步下降,但甲烷的吸附量卻稍有增加,說明活性炭的比表面積不是影響甲烷吸附量的唯一因素。
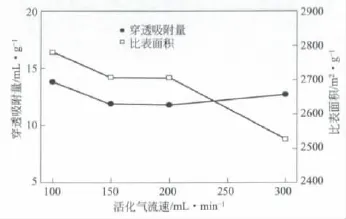
圖6 活化氣流速不同對甲烷吸附性能及活性炭比表面積的影響
2.4 CMC添加量不同對活性炭吸附性能的影響
實驗室自制的石油焦基活性炭為粉末狀固體,無實際工業(yè)應(yīng)用價值,CMC以其價格便宜、后處理溫度較低而成為一種常用的黏結(jié)劑。圖7是堿焦比為4時成型前后活性炭的甲烷吸附穿透曲線,成型后活性炭的抗壓碎強度如表3所示。從圖7和表3可以看出,添加質(zhì)量分數(shù)為10%CMC成型的活性炭其甲烷穿透吸附量略低于粉末活性炭,但其飽和吸附量無明顯差異。當(dāng)CMC添加量較低時,活性炭的抗壓碎強度無法保證;當(dāng)CMC的含量進一步增加時,黏結(jié)劑會堵塞活性炭的微孔,造成吸附量的下降,所以10% (質(zhì)量分數(shù))的添加量是較為理想的比例。
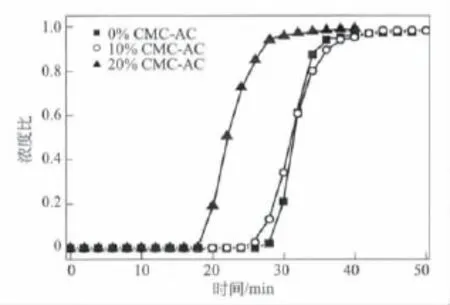
圖7 成型前后活性炭的甲烷穿透曲線

表3 添加不同CMC成型后活性炭的抗壓碎強度
2.5 石油焦基活性炭的再生性能
再生性能是評價吸附劑好壞的一個重要指標(biāo),良好的再生性能可以有效降低工業(yè)生產(chǎn)成本。活性炭的再生方法有很多,本文選擇熱處理的方法,將吸附飽和的活性炭在馬弗爐中200℃處理3 h后進行再生性能的考察。
圖8是再生活性炭對甲烷的穿透吸附量和飽和吸附量的柱狀圖。從圖8可知,成型后的活性炭經(jīng)過5次再生后,其穿透吸附量及飽和吸附量都有所降低,這與吸附過程的微孔填充及中孔毛細凝聚現(xiàn)象有關(guān),但未出現(xiàn)吸附量明顯降低的現(xiàn)象。說明成型活性炭的再生性較好。
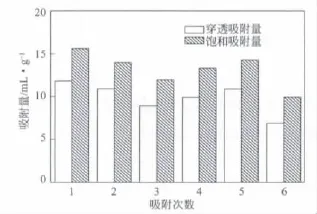
圖8 再生次數(shù)對條狀活性炭吸附量的影響
3 結(jié) 論
(1)石油焦預(yù)處理正交實驗結(jié)果表明,在考慮的4種因素中,對活性炭的比表面積影響大小的順序為:改性時間>液固比>改性溫度>氧化劑種類,高比表面積活性炭的最佳制備條件是:H2O2為氧化劑,液固比為30 m L/g,改性時間為6 h,改性溫度為60℃。
(2)在活性炭的制備過程中,采用雙氧水預(yù)處理石油焦可以改變其表面性質(zhì),利于活化的進行,使甲烷的吸附性能提高36%左右;活化過程中氣體流速的變化會降低活性炭的比表面積,但不會對甲烷的吸附量造成明顯影響,說明比表面積不是影響甲烷吸附量的唯一因素;原料粒度過細使活性炭孔道結(jié)構(gòu)更發(fā)達,對甲烷的吸附力變?nèi)酰焕诩淄榈奈健?/p>
(3)添加10% (質(zhì)量分數(shù))羧甲基纖維素鈉成型后的石油焦基活性炭既保證了良好的力學(xué)強度,又保證了甲烷的吸附量;且多次使用后,吸附量雖稍有下降,但穩(wěn)定性較好。
[1] 李元建.中國煤層氣產(chǎn)業(yè)開發(fā)利用現(xiàn)狀與對策分析 [J].中國礦業(yè),2010,19(6):8-9.
[2] 辜敏,鮮學(xué)福,張代均,等.變壓吸附技術(shù)分離CH4/N2氣體混合[J].煤炭學(xué)報,2002,27(2):197-199.
[3] 趙紅濤,徐偉,吳建軍,等.煤層氣綜合利用現(xiàn)狀及前景[J].能源技術(shù)與管理,2011(4):105.
[4] 雷利春.煤礦乏風(fēng)中低濃度甲烷的變壓吸附提純 [D].大連:大連理工大學(xué),2010.
[5] 黃格省,于天學(xué),李雪靜.國內(nèi)外煤層氣利用現(xiàn)狀及技術(shù)途徑分析[J].石化技術(shù)與應(yīng)用,2010,28(4):342-344.
[6] 陶鵬萬,王曉東,黃建彬.煤層氣低溫分離提濃甲烷工藝:中國,200410040155.4[P].2006-01-11.
[7]Makaruk A,Miltner M,Harasek M.Membrane biogas upgrading processes for the production of natural gas substitute[J].Separation and Purification Technology,2010,74(1):83-92.
[8] 曾征.淺析變壓吸附氣體分離的技術(shù)及應(yīng)用 [J].科技資訊,2005,14(3):5.
[9] 王琰.空氣中低含量甲烷的變壓變壓吸附分離 [D].天津:天津大學(xué),2004.
[10] 張曉環(huán).低濃度甲烷的吸附研究 [D].上海:華東理工大學(xué),2012.
[11]Yuan B,Wua X F,Chen Y X,et al.Adsorptive separation studies of ethane-methane and methane-nitrogen systems using mesoporous carbon[J].Journal of Colloid and Interface Science,2013,394:445-450.
[12]Liu C M,Dang Y Y,Zhou Y P,et al.Effect of carbon pore structure on the CH4/N2separation[J].Adsorption,2012,18:321-325.
[13]張建.新型CO2-PSA吸附劑的研制與應(yīng)用研究[D].大連:大連理工大學(xué),2002.
[14]Quinn D F,MacDonald J A.Natural gas storage[J].Carbon,1992,30(7):1997.
[15] 孫艷,周理.氣體在多孔材料中的儲存 [J].油氣儲運,2009,28(2):17-19.
[16] 蔣寶城.高比表面積活性炭的制備及其初步應(yīng)用 [D].大連:大連理工大學(xué),2010.
[17]Jiang B C,Zhang Y C,Zhou J X.Effects of chemical modification of petroleum cokes on the properties of the resulting activated carbon[J].Fuel,2008,87(10-11):1844-1848.
猜你喜歡
紡織科學(xué)研究(2020年1期)2020-05-21 00:31:06
中國塑料(2016年12期)2016-06-15 20:30:07
中國塑料(2016年2期)2016-06-15 20:30:00
中國塑料(2016年2期)2016-06-15 20:29:59
中國塑料(2016年5期)2016-04-16 05:25:36
廣西林業(yè)科學(xué)(2016年3期)2016-03-16 05:43:30
中國塑料(2015年3期)2015-11-27 03:41:38
中國塑料(2015年11期)2015-10-14 01:14:14
中國塑料(2015年9期)2015-10-14 01:12:17
中國塑料(2015年4期)2015-10-14 01:09:19
