EPDM基三元共聚高吸油樹脂的合成
2012-12-23 09:00:00周曉明亢進現揣成智
合成樹脂及塑料 2012年2期
周曉明,亢進現,揣成智
(天津科技大學材料科學與化學工程學院,天津市 300457)
EPDM基三元共聚高吸油樹脂的合成
周曉明,亢進現,揣成智
(天津科技大學材料科學與化學工程學院,天津市 300457)
采用懸浮聚合法,以三元乙丙橡膠(EPDM)為大分子單體,以不含極性基團的烯烴分子苯乙烯、含不飽和側基的衣康酸為共聚單體進行三元共聚合,得到一種新型高吸油樹脂。采用傅里葉變換紅外光譜、熱重分析、掃描電子顯微鏡等手段對樹脂的結構、吸油性能及吸油后斷面的微觀形貌進行了表征。加入適量的衣康酸共聚單體可以大幅提高EPDM基吸油樹脂對柴油的吸收能力,對柴油最大吸油倍率可達19.8 g/g;同時還可顯著提高吸油樹脂的吸油速率;通過掃描電子顯微鏡觀察可推斷,吸油樹脂在聚合過程中形成了互穿網絡結構。
吸油樹脂 三元乙丙橡膠 懸浮聚合 互穿網絡
與傳統吸油材料相比,高吸油樹脂具有與高吸水性聚合物基本相同的三維網狀結構,具有吸油種類多、速度快,吸油時不吸水、回收方便等優點,用途廣泛。根據使用的合成單體可將吸油樹脂分為丙烯酸酯類[1-3]和烯烴類[4-5],其中,烯烴類吸油樹脂包括以三元乙丙橡膠(EPDM)為單體制備的合成樹脂。
EPDM基吸油樹脂[6]通常以含不飽和基團的EPDM橡膠為大分子反應單體,以不含極性基團的烯烴分子為共聚單體,加入適量引發劑、交聯劑、分散劑等經懸浮聚合形成適度交聯的網絡結構,吸收的油以范德華力保存于網絡中。該吸油樹脂吸油倍率較高,保油性能好且經溶劑萃取后可重復使用。但因EPDM基吸油樹脂在合成過程中存在易黏結及對溫度變化敏感等問題,限制了其發展和應用。為進一步提高吸油樹脂的吸油能力,改善吸油樹脂表面結構,減少交聯網絡的空間位阻,克服其吸油倍率低和吸油速率慢等缺陷,本工作從分子設計的角度出發,選擇柔性大分子EPDM、苯乙烯(St)為單體,引入含不飽和側基的衣康酸(ITA)為共聚單體進行三元共聚合,得到高性能吸油樹脂,研究了吸油樹脂的吸油性能及微觀結構。
1 實驗部分
1.1 原料
EPDM,香港創達塑膠原料有限公司生產;St,天津大茂化學試劑廠生產;ITA,上海試劑廠生產;二乙烯基苯(DVB),成都科龍化工試劑廠生產;過氧化苯甲酰(BPO),上海天蓮精細化工有限公司生產;甲苯、環己烷,均為天津北方天醫化學試劑廠生產;明膠、磷酸三鈣,均為天津永大化學試劑開發中心生產。以上化學試劑均為分析純。
1.2 儀器
Vector22型傅里葉變換紅外光譜儀,德國Bruker公司生產。Q500型熱重分析儀,美國TA儀器公司生產。JSM-6380LV型掃描電子顯微鏡,日本JEOL公司生產。
1.3 EPDM基三元共聚吸油樹脂的合成
采用懸浮聚合法,在N2保護下按比例稱取定量的去離子水及分散劑(明膠和磷酸三鈣),于40℃下攪拌5 min;然后加入ITA繼續攪拌5 min;在N2保護下加入計量的EPDM,St,BPO,DVB后劇烈攪拌并升溫至80℃,反應8 h;反應結束后冷卻、過濾,用稀鹽酸洗除未反應的單體及分散劑;產物經萃取后在60℃真空干燥24 h,得到白色顆粒狀化學交聯共聚物。反應過程見式(1)。
1.4 吸油倍率的測定
稱重法:準確稱取吸油樹脂置于盛有柴油的燒杯中,定時將樹脂從燒杯中取出,用濾紙擦去表面浮油,然后稱重。吸油倍率可按Q=(ms-md)/md計算,式中:Q為吸油倍率,md,ms分別表示吸油前、后樹脂的質量。

熱重(TG)分析:在N2保護下,用熱重分析儀測試吸油樹脂吸油前后的失重行為。試樣質量一般為2~5 mg;升溫速率為10℃/min;N2流量為40 mL/min。采用TG曲線作圖法計算吸油倍率。
2 結果與討論
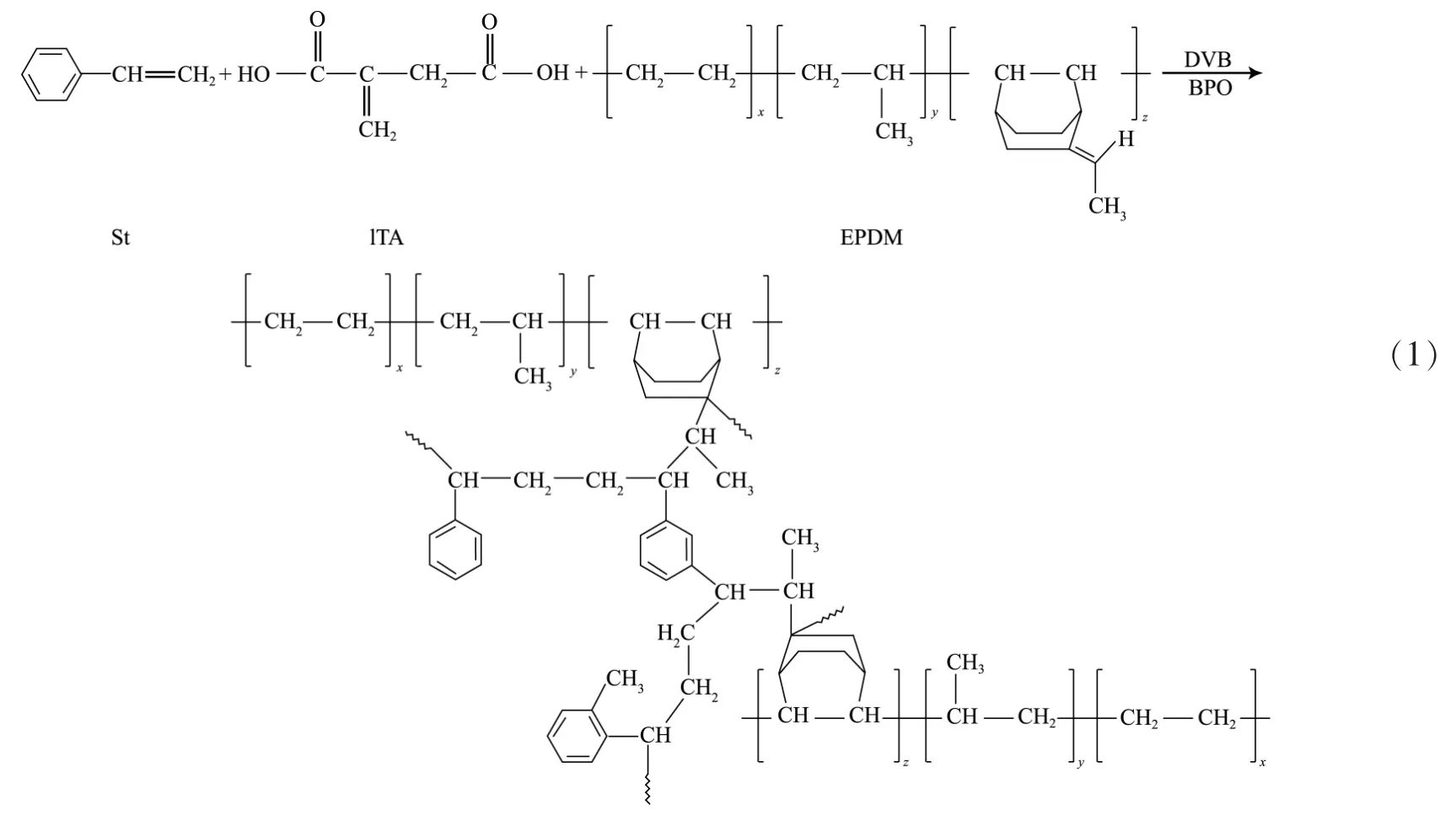
2.1 反應物組成對吸油性能的影響
合成EPDM基三元共聚高吸油樹脂的配方見表1。配方A,B,C,D,E各組均得到了顆粒大小不同的吸油樹脂,其中,A組產品呈乳白色,B組呈淺黃色,C組、D組和E組呈透明狀,顏色稍微發黃。測試表明:E組顆粒最小,對柴油的吸油倍率最高;而B組顆粒最大,對柴油的吸油倍率最低。主要原因在于B組配方中St反應單體被ITA全部取代后,EPDM自聚合傾向增加,形成的樹脂網絡結構中缺少剛性,網絡結構容易坍塌,導致吸油倍率陡降。配方中ITA組分一方面作為分散劑分散于水中;另一方面也作為反應單體參與共聚合或發生自聚合后同EPDM基吸油樹脂形成互穿網絡。因此,加入ITA明顯改變了吸油樹脂的網絡結構,使吸油樹脂的吸油倍率和吸油速率均顯著變化。
由表1及圖1看出:當m(St)/m(ITA)為1∶3時,得到的吸油樹脂(E組)對柴油的吸油倍率最高。B組吸油倍率最小,在4天內即達到飽和吸油狀態;而吸油倍率較高的C組和E組在8天才達到飽和狀態;在1~4天,C,D,E組的吸油速率較快,而ITA含量較高的B組吸油速率最慢,A組次之。這表明配方中適量加入ITA可以提高吸油樹脂的吸油速率。
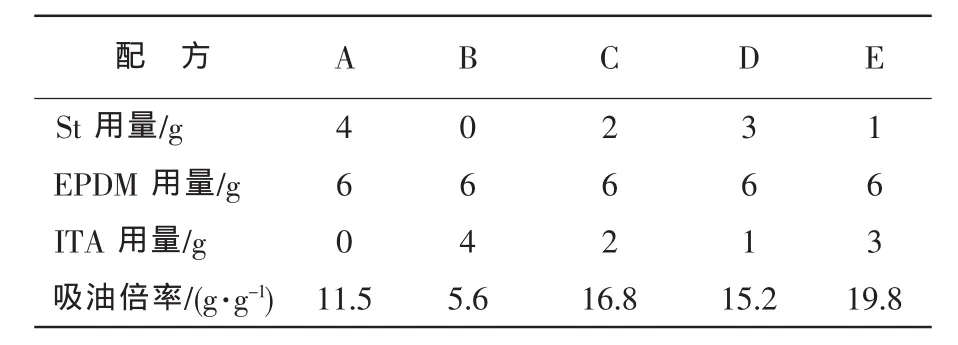
表1 吸油樹脂的配方及吸油倍率Tab.1Formulae and oil absorptivity of the oil-absorbing resin
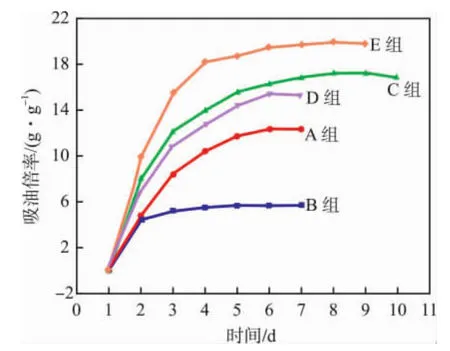
圖1 吸油樹脂的飽和吸油時間曲線Fig.1 Curves of saturation oil absorption time of the oil-absorbing resin
2.2 吸油樹脂的結構表征
由圖2可以看出:除包含EPDM特征官能團的紅外振動吸收峰外,1 720 cm-1處為羰基的伸縮振動峰;1 156,1 046 cm-1處為C—O伸縮振動吸收峰,共同驗證了酯基的存在,表明ITA參與了共聚合。此外,在1 602 cm-1處出現苯環上C=C骨架伸縮振動峰,906~1 069 cm-1出現苯環上C—H面外振動峰,同時在1 640~1 675 cm-1未出現明顯的C=C—H及C=CH2振動吸收峰,表明共聚物中St與ITA上的不飽和基團均發生了接枝交聯,形成了一定的網絡結構。

圖2 EPDM基三元共聚高吸油樹脂的傅里葉變換紅外光譜Fig.2 FTIR spectra of the high oil-absorbing resin terpolymerized from EPDM-St-ITA
2.3 吸油樹脂的TG分析
由圖3中吸油前曲線可知:在N2保護下,制備的高吸油樹脂在450℃時才開始大量分解,低于300℃時不分解。由此說明,該高吸油樹脂在通常的使用溫度范圍內是極其穩定的,可以正常使用。吸油后的曲線顯示了三階段的分解過程:第一階段是脫柴油階段,發生在50~300℃;第二階段是穩定階段,發生在300~450℃,該階段柴油已經完全脫除;從第三階段開始為高吸油樹脂的分解階段。通過在TG曲線作圖法測得樹脂吸油倍率為20.1 g/g,同稱重法測得數據基本一致。
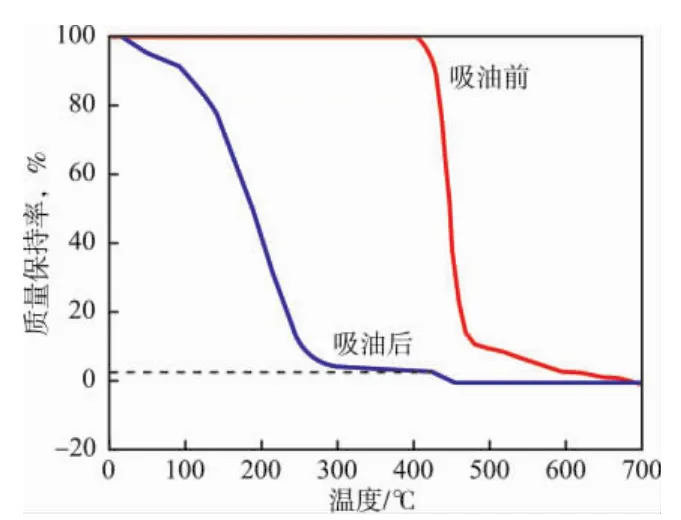
圖3 吸油樹脂吸油前后的TG分析Fig.3 TG curves of the oil-absorbing resin before and after absorbing oil
2.4 吸油樹脂微觀結構分析
由圖4看出:吸油樹脂吸油后內部被柴油充滿,形成類似浮雕的互穿網絡形態結構。該結構中凸起部分可能為網絡結構中的空隙部分;當樹脂與柴油接觸時,吸油樹脂分子內的親油基鏈段和油分子的溶劑化作用使樹脂膨潤,柴油逐漸滲透到樹脂內部,占據網絡結構空間,直至達到溶脹平衡。E組吸油樹脂在吸油前網絡結構較為松散,內部空間較大,可容納更多柴油;而C組吸油樹脂網絡較為緊密,對油品容納空間較小,吸油倍率相對E組較低。

圖4 吸油樹脂吸油后淬斷面掃描電子顯微鏡照片(×2 000)Fig.4 SEM photos of cross section of the oil-absorbing resin after absorbing oil
3 結論
a)采用懸浮聚合法制備了EPDM基三元共聚高吸油樹脂。
b)m(EPDM)/m(St)/m(ITA)為6∶1∶3時,得到的吸油樹脂對柴油具有較高的吸油倍率,且在短時間內吸油速率較快。
[1]藺海蘭,廖建和,廖雙泉,等.丙烯酸酯系共聚物高吸油樹脂的合成及性能研究[J].彈性體,2006,16(5):34-39.
[2]王勇,趙彥芝,萬濤.丙烯酸系二元共聚高吸油性樹脂的合成及性能研究[J].功能材料,2004,35(5):638-640.
[3]孫曉然,張秀玲.丙烯酸酯-苯乙烯共聚物高吸油樹脂的合成與性能[J].塑料工業,2003,31(7):7-13.
[4]吳波,周美華.高吸油樹脂[J].現代塑料加工應用,2006,18 (2):62-64.
[5]劉秀齊,孫國,李海東,等.礦粉/EPDM新型吸油材料的制備及其吸油性[J].彈性體,2007,17(5):49-52.
[6]Zhou Xiaoming,Chuai Chengzhi.Synthesis and characterization of a novel high-oil-absorbing resin[J].Journal of Applied Polymer Science,2010,115(6):3321-3325.
TG 324
B
1002-1396(2012)02-0009-04
2011-10-02。
2011-12-30。
周曉明,1974年生,博士,副研究員,2006年畢業于吉林大學高分子專業,現從事功能高分子材料和可降解高分子材料研究。聯系電話:13920884902;E-mail:xiaomingzhou@tust.edu.cn。
(編輯:王蕾)
