核轉錄因子kappaB在急性心肌梗死后心室重塑中作用研究
2012-11-21 02:28:44陜西省寶雞市中心醫院寶雞721008樊冬梅
陜西醫學雜志 2012年12期
陜西省寶雞市中心醫院(寶雞721008) 樊冬梅 薛 莉
心肌梗死后普遍伴隨著左心室重塑,指的是左室腔、幾何結構和功能的改變。左心室重塑是一個慢性進展過程,心肌細胞和細胞外基質遞進性改變,導致左室的擴大、收縮功能的下降和潛在的室性心律失常、心衰、和隨后心血管性猝死[1]。心室重塑過程的具體機制不斷得到闡明,其中炎癥反應和其細胞因子的作用越來越受到重視。本研究應用細胞免疫化學染色觀察急性心肌梗死(AMI)后心室重塑患者外周血單個核細(PBMCs)的核因子-kappaB(NF-κB)活性及其與血漿細胞因子腫瘤壞死因子α(TNF-α)、白介素-1β(IL-1β)分泌的相關性來探討心室重塑的發病機制。
1 資料與方法
1 一般資料 急性心肌梗死患者來自我院2008年12月至2009年9月心內科住院病人,共58例,AMI的診斷標準根據1981年WHO制定心肌梗死診斷標準。心功能采取killip分級法,分為Ⅰ、Ⅱ、Ⅲ、Ⅳ級。所有受試者均除外陳舊性心肌梗死、其他心臟病、各種炎癥、惡性腫瘤、風濕活動、內分泌疾病等。以左心室舒張末期容積指數增加率(ΔLVEDVI)>20%作為AMI后心室重塑的分組標準[2]:A組:左心室重塑組(ΔLVEDVI>20%)33例,男20例,女13例,平均年齡為59.7±8.4;B組:非左心室重塑組(ΔLVEDVI≤20%)25例,男15例,女10例,平均年齡為58.6±5.9。另取門診體檢健康者為C組:20例,男性12例,女性8例,平均年齡57.8±6.5歲。
2 方 法
2.1 試劑及儀器 :淋巴細胞分離液(中國醫學科學院生物工程研究所)、1640培養基(Sigma Co)、小牛血清(FCS)(鄭州佰安生物工程有限公司)、細胞離心機cytospin-2型(British.SHANDON)、NF-kB(p50)兔抗、鏈酶親和素一生物素過氧化物酶試劑盒(SABC),試劑盒(單克隆鼠抗兔)及DAB試劑盒均購自武漢博士德公司。TNF-α、IL-1β放免試劑盒購自解放軍總醫院科技開發中心放射免疫研究所。
2.2 標本采集:采血均得到患者同意,用肝素鋰5ml真空采血管無菌采取確診AMI患者發病的3d及14d空腹靜脈血5ml。
2.3 外周血單個核細胞(PBMCs)的分離:取抗凝3ml血液用磷酸緩沖液(PBS)1∶1稀釋后,以2∶1體積置于淋巴細胞分離液上,3000r/min離心30min,收集試管中間懸浮的一層乳白色的液體,即為外周血單個核細胞(PBMCs)。再以磷酸緩沖液(PBS)洗滌3次,每次1000r/min離心5min。用臺盼藍拒染試驗活細胞比例大于95%為有效標本。1640培養基調整細胞數為25×107個/ml,取100ul細胞混懸液滴入細胞離心機600r/min,離心6min,使細胞甩于載玻片上,室溫下自然風干,丙酮固定5min,干燥后保存于-70℃冰箱待測NF-κB活性。
2.4 NF-κB的免疫組化染色(SABC法):第一抗體選用兔抗NF-κBp50亞單位的多克隆抗體,抗體稀釋度經預實驗定為1×100,二抗為生物素化單克隆小鼠抗兔IgG,采用SABC法試劑盒檢測。操作步驟按試劑盒說明書進行,每張涂片隨機選取5個不重疊的高倍鏡視野,隨機計數500個細胞,計算細胞核陽性的細胞數占細胞總數的百分率。免疫細胞化學染色結果判定:PBS代替一抗為空白對照,細胞染色為棕黃色者為陽性,定位于胞質與胞核。PBMCs中NF-κB的活化水平用NF-κB(+)cells占所有定量化的PBMCs的百分比來表達。即陽性率=陽性細胞數/總細胞數×100%
2.5 血漿細胞因子的測定:取出2ml血液置于離心管中,以2500r/min離心20min,取上層血漿置于-70℃冰箱保存,待測細胞因子(TNF-α,IL-1β);利用液相競爭抑制原理,采用平衡法對樣品進行測定。將待測樣品或標準與限量的抗血清及標記的抗原加在一起進行競爭結合反應,加入免疫分離劑分離出抗原抗體復合物,測定復合物的放射性(B),計算各標準管的結合率(B/B0%.)做出標準曲線,由GC-1200r放射免疫計數器查出樣品濃度。
2.6 超聲心動圖檢查:所有患者入院后第3天和第30天進行超聲心動圖檢查,測定左心室舒張末期內徑(LVED)。按Simpson法計算左心室舒張末期容積(LVEDV),經體表面積校正后計算出左心室舒張末期容積指數(LVEDVI),并進一步計算出ΔLVEDVI =(30dLVEDVI-3dLVEDVI) /3dLVEDVI×100%。
2.7 統計學處理:應用SPSS 11.5分析軟件,計量資料均采用±s表示,計數資料用χ2檢驗,三組間細胞因子比用單因素方差分析;PBMC的NF-κB與細胞因子間的相關程度采用直線相關性分析法;進行統計處理,顯著性檢驗水準取a=0.05。
結 果
1 NF-κB檢測結果 AMI后重塑組血漿TNF-α、IL-1β和外周血單個核細胞NF-κB核染色陽性百分率測量結果均顯著高于未重塑組、健康對照組(見表1及附圖)。AMI后重塑患者外周血單個核細胞NF-κB核染色陽性百分率為顯著高于未重塑組、健康對照組3d三組分別:26.0%±7.60%,22.30%±5.46%、10.05%±5.23%;14d:20.45%±5.46%、13.00%±4.62% 、10.0%±5.23%。
2 通過相關性分析發現 急性心肌梗死患者外周血單個核細胞NF-κB核染色陽性百分率與TNF-α、IL-1β表達均呈正相關(rTNF-α=0.649,P<0.01;rIL-1β=0.535,P<0.01)(見表2)。
3 通過χ2檢驗發現 對照組和觀察組的性別和心功能分級(killip分級),及合并癥無顯著性差異。
表1 外周血單個核細胞NF-kB染色陽性率與血漿細胞因子測定(%,ng/ml,±s)
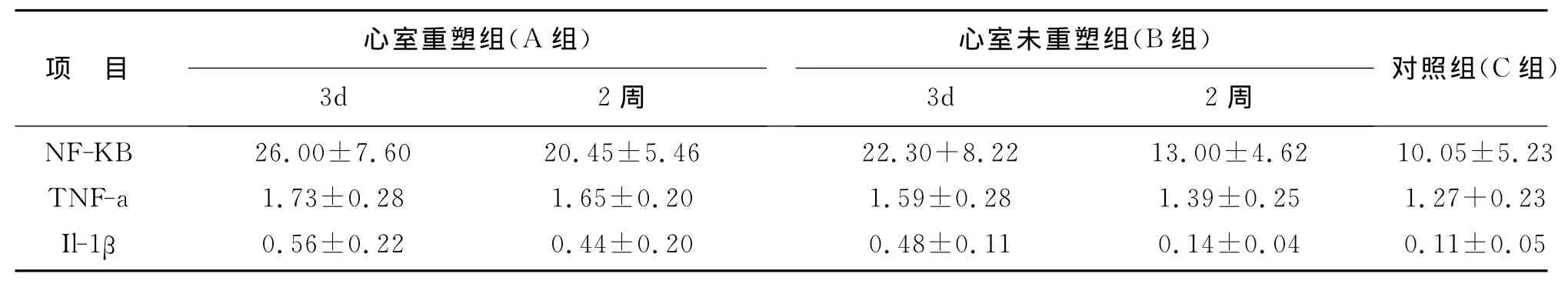
表1 外周血單個核細胞NF-kB染色陽性率與血漿細胞因子測定(%,ng/ml,±s)
注:在發病3d,A組與B組方差分析值P>0.05.;14d的A和B圖方差分析值P>0.05.
心室重塑組(A組) 心室未重塑組(B組)項 目對照組(C組3d 2周3d 2周NF-KB TNF-a Il-1β)0.11±0.05 26.00±7.60 1.73±0.28 0.56±0.22 20.45±5.46 1.65±0.20 0.44±0.20 22.30+8.22 1.59±0.28 0.48±0.11 13.00±4.62 1.39±0.25 0.14±0.04 10.05±5.23 1.27+0.23

表2 NF-KB 相關性分析
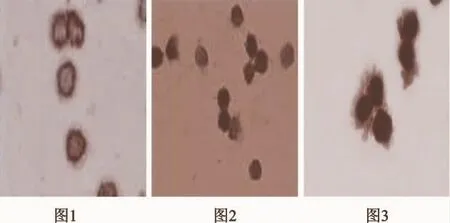
圖1、2:AMI后未重塑患者;圖3:AMI后重塑患者;圖:外周血PBMCs中NF-kB免疫細胞化學染色結果(10×100倍)
討 論
急性心肌梗死心肌壞死可繼發的局部和全身的系統性炎癥反應導致組織損傷和心肌擴大及心力衰竭,即不可逆的心室重塑[3]。
NF-κB扮演一個炎癥反應和先天免疫的中樞調節因子,NF-κB的活性是通過控制細胞的增殖遷移、細胞的分化和細胞的凋亡發揮作用的。它屬于Rel家族(p50,p52,p65,c-Rel,and RelB),享有保守的Rel同源序列,能形成異源二聚體(p50/p65,p50/p50,或p65/p65),存在于細胞質中。在靜息狀態下,細胞質中NF-κB是靜止的、無活性的,被IκB抑制。各種刺激包括致炎因子,氧化應激,細菌和病毒產物,或缺氧刺激激活。在NF-κB經典的激活通路中,IKK復合物磷酸化IκB,導致26S蛋白體泛蛋白化和降解,因此暴露NF-κB亞單位上的核定位信號和NF-κB的轉錄上調各種靶基因,參與各種生理和病理過程,包括AMI。AMI發生后,激活的NF-κB介導了左心室重塑和心功能惡化[4]。事實上,對于NF-κB功能更詳細功能是不同的亞單位有不同的功能,在這項研究,我們的焦點集中在心梗后NF-κB p50,p50亞單位是通常考慮是NF-κB復合物的抑制劑[5],因為p50/p50同源二聚體是一直轉錄活性的必需的。
許多研究顯示NF-κB是心肌缺血的炎癥反應介質,TOLL受體,它的信號傳導通過NF-κB,而TOLL受體在心肌梗死后左室重塑起中樞性的作用[6,7],通過用IKB酶的磷酸化抑制劑抑制NF-κB的核易位,左室重塑和心力衰竭可以被改善[8],注射有高親和力雙股誘導DAN的阻斷NF-κB,可以減輕大鼠模型的心肌缺血和再灌注損傷[9],Frantz等[10]用 ELISA 等方法對心肌梗死后10周鼠的心肌細胞NF-κB易位情況的研究表明:在心肌梗死后心肌細胞中NF-KB慢性并持續的激活。動物實驗表明NF-κB的活化可以促發并加劇心肌肥厚、心功能惡化[11]。NF-κB能發揮細胞保護性的作用,在心肌梗死后心室重塑和心力衰竭中也發揮調節作用[12]。因此阻斷NF-κB活性被認為是一個有前景的阻止有害的心梗后左室重塑途徑。
急性心肌梗死后在體內的IL-1β表達是增加的,心肌壞死部位直接產生白介素-1β炎性因子,負責內皮系統的激活、白細胞聚集和炎癥反應擴大化;IL-1β也可導致非心梗的部位心肌損害[13];并可促進心肌纖維化和心肌肥大[14,15]。相應地,TNF-a也是增加的,持續存在的TNF-a導致心肌細胞的表型轉變和MMPs活性增加,不斷增加的TNF-a,抑制心肌收縮力、誘發心肌細胞凋亡和基質纖維增生,加劇了心室重塑的進程[16]。
本實驗研究表明:正常人PBMC中就有NF-κB的微量表達,NF-κB染色多位于胞質中,核染色較少,已證實適量的NF-κB表達對人體組織的生長和維持內環境穩定起重要的調節作用。AMI患者的外周單個核細胞的NF-κB表達在3d明顯高于正常人(見圖),且心室重塑組與未重塑組無差別。這表明,在急性心肌梗死后大量炎癥因子的刺激下,NF-κB與IκB解離,發生了核移位,促使NF-κB的過度表達。14d時,AMI后心室重塑組的患者NF-κB顯著高于未重塑患者,兩者比較有統計學意義,說明NF-κB在AMI后持續活化激活參與了心室重塑發生發展;未重塑組雖已下降,高于正常對照組,但已無統計學差異;另外,心室重塑組血漿細胞因子TNF-α、IL-1β的水平較心室未重塑組、對照組顯著升高,并且NF-κB染色的陽性百分率與細胞因子 TNF-α、IL-1β的水平顯著正相關,rTNF-α=0.649,P<0.01;rIL-1β=0.535,P<0.01(見表2)。心室未重塑組14d和對照組,核因子與腫瘤因子、白介素相關性較心室重塑組低,或無相關性(見表2);這說明心室重塑組激活轉錄水平的NF-kB可以促進靶基因細胞因子TNF-α、IL-1β分泌來參與心室重塑及心力衰竭等病理生理過程。
另外本實驗還發現,58例急性心肌梗死患者,行PCI者35例;發生左心室構(LVRM)的13例,未行冠脈介入術(PCI)者23例;發生LVRM的18例;經卡方檢驗χ2=9.431,P=0.002,P<0.01。所以兩組重塑發生率有差別。說明行盡早行PCI、恢復冠脈重建,有可能減少急性心肌梗死后心室重塑的發生率。
本實驗采用細胞免疫細胞化學法,比較直觀,可以清楚觀察NF-κB p50,在胞質和細胞核內分布的情況,操作簡便,可同時對NF-κB進行定位和半定量檢測。但若對NF-κB精確地檢測采用Western blont或EMSA法。
[1]St John Sutton M,Lee D,Rouleau JL et al.Left.ventricular remodeling and ventricular arrhythmiasafter myocardial infarction.Circulation.2003,107:2577–2582.
[2]Bolognese L,Neskovic AN,Parodi G,et al.Left ventricular remodeling after primary coronary angioplasty:patterns of left ventricular dilation and long-term prognostic implications.Circulation,2002;106:2351–2357.
[3]Frangogiannis N.The immune system and cardiac repair.Pharm Res,2008,58:88–111.
[4]Onai Y,Suzuki J,Maejima Y,et al.Inhibition of NF-{kappa}B improves left ventricular remodeling and cardiac dysfunction after myocardial infarction.Am J Physiol Heart CircPhysiol.2007,292:H530– H538.
[5]Driessler F,Venstrom K,Sabat R,et al.Molecular mechanisms of interleukin-10-mediated inhibition of NF-kappaB activity:a role for p50.Clin Exp Immunol,2004,135:64–73.
[6]Timmers L,Sluijter JP,Van–Keulen JK,et al.Tolllike receptor 4mediates maladaptive left ventri-cular remodeling and impairs cardiac function after myocardial infarction.Circ Res,2008,102:257–264.
[7]Shishido T,Nozaki N,Yamaguchi S,et al.Toll-like receptor-2modulates ventricular remodeling after myocardial infarction.Circulation,2003,108:2905–2910.
[8]Onai Y,Suzuki J,Maejima Y,et al.Inhibition of NF-{kappa}B improves left ventricular remodeling and cardiac dysfunction after myocardial infarction.Am J Physiol Heart CircPhysiol,2007;292:H530– H538.
[9]Morishita R,Sugimoto T,Aoki M,et al.In vivo transfection of cis element“decoy”against nuclear factor-kappaB binding site prevents myo-cardial infarction.Nat Med,1997,3:894–899.
[10]Frantz S,Fraecarllo D,Wagner H,et al.Sustained activation of nuclear factor kappa B and activator protein 1in chronic heart failure[J].Cardiovasc,Res,2003,57:749-756.
[11]Gupta S,Young D,Sen S.Inhabition of NF-KappaB Induces regression of cardiac hypertrophy,independent of blood pressure control,in spontaneously hypertensive rats[J].Physiol Heart CirC Physiol,2005,289(l):H17-9.
[12]Misra A,Haudek SB,Knuefermann P,et al.Nuclear factorkappaB protects the adult cardiac myocyte against ischemia-inducedapoptosis in a murine model of acute myocardial infarction.Circulation,2003,108:3075–3078.
[13]Dinarello C.Immunological and inflammatory functions of the interleukin-1family.Annu Rev Immunol 2009,27:519–550.
[14]Bujak M,Dobaczewski M,Chatila K,et al.Interleukin-1receptor type I signaling critically regulates infarct healing andcardiac remodeling.Am J Pathol,2008,173:57–67.
[15]Abbate A,Salloum F,Vecile E,et al.A.Anakinra,a recombinant human interleukin-1receptor antagonist,inhibits apoptosis in experimental acute myocardia infar ction.Circulation,2008,117:2670–2683.
[16]Sun M,Dawood F,Wen WH,et al.Excessive tumor necrosis factor activation after infarction contributes to susceptibility of myocardial rupture and left ventricular dysfunction.Circulation,2004,110:3221–3228.
