Myceliothermophin E片斷(Z)-5-(2-甲基亞丙基)-3-吡咯啉-2-酮的合成及其Baylis-Hillman模型反應
2012-11-21 01:09:54曲世偉
合成化學 2012年5期
關鍵詞:方法
金 瑛, 曲世偉
(1. 吉林醫藥學院,吉林 吉林 132013; 2.北京大學 深圳研究生院,廣東 深圳 510085)
天然產物Myceliothermophin E是由Shih-Hsiung Wu[1]從嗜熱菌中分離得到的聚酮類化合物,具有明顯的抗癌活性,分子內含有(Z)-5-(2-甲基亞丙基)-3-吡咯啉-2-酮(1)與八氫萘環(2, Chart 1)兩個主要片斷,共含有5個手性中心、4個雙鍵。從逆合成分析原理考慮,首先分別合成1和2; 再經Baylis-Hillman反應由1與2合成Myceliothermophin E。

Chart 1

方法一


方法二
Scheme1

Scheme2
目前已分離得到多種含3-吡咯啉-2-酮結構的聚酮類化合物[2~10],根據對類似分子生物活性的研究表明,3-吡咯啉-2-酮結構是生物活性的必需結構。不同天然產物中3-吡咯啉-2-酮片斷有著明顯差異,其合成方法各異[5~9]。3-吡咯啉-2-酮片斷的成功合成是Myceliothermophin E全合成的關鍵所在。
本文選擇了Myceliothermophin E片斷(Z)-1作為研究對象,1的合成方法未見相關報道。本文采用兩種方法合成了1(Scheme 1)。方法一是以鏈狀化合物5為原料通過改良的HWE反應制備順式α,β-不飽和酯6;再經分子內親核取代反應進行環合、消除反應制得(Z,E)-1;方法二則以吡咯為原料,經Mukaiyama aldol反應引入C5-位取代基,再經過消除反應制得(Z)-1。
(E)-11與無水乙醛進行Baylis-Hillman反應(Scheme 2),為Myceliothermophin E的全合成提供了一個可能的模型反應。
1 實驗部分
1.1 儀器與試劑
Perkin-Elmer 351型旋光儀;Brüker Avance 300(500)型核磁共振儀(CDCl3為溶劑,TMS為內標);ESI-HR-MS型質譜儀。
二氯甲烷、乙腈采用CaH2回流純化;THF和Et2O采用金屬鈉回流純化;薄層層析采用GF254高效板;快速柱層析用硅膠(200目~300目),青島海洋化工廠;其余所用試劑均為分析純。
1.2 1的合成
(1) 方法一

在反應瓶中加入754 mg(0.2 mmol)的TFA(2 mL)溶液,攪拌下于50 ℃反應2 h。冷卻至0 ℃,加入乙酸乙酯(15 mL)與飽和NaHCO3溶液(15 mL)淬滅反應,用水(10 mL)洗滌,無水Na2SO4干燥,減壓蒸除溶劑后經硅膠柱層析(洗脫劑:A=1 ∶3)分離得無色油狀液體(Z,E)-1 23 mg,產率84%;1H NMRδ: 9.12(s, 1H), 6.90(dd,J=1.4 Hz, 5.5 Hz, 1H), 6.13(dd,J=0.6 Hz, 5.4 Hz, 1H), 5.14(d,J=9.8 Hz, 1H), 2.79~2.71(m, 1H), 1.75(s, 1H), 1.12(d,J=6.6 Hz, 1H);13C NMRδ: 173.3, 139.1, 136.7, 124.9, 123.8, 27.8, 22.9; HR-ESI-MSm/z: Calcd for C8H12NO+{[M+H]+} 137.084 1, found 137.085 3。
(2) 方法二
在反應瓶中將95.8 g(19 mmol)溶于Et2O(200 mL),降溫至-80 ℃,氮氣保護,攪拌下依次加入異丁醛4.1 g(57 mmol)的Et2O(30 mL)溶液和SnCl43 mL(25 mmol)的CH2Cl2(20 mL)溶液,于-80 ℃反應4 h。加入飽和NaHCO3溶液(50 mL)淬滅反應,慢慢升至室溫,用乙酸乙酯(3×30 mL)萃取,合并有機相,用無水Na2SO4干燥,減壓蒸除溶劑后經硅膠柱層析(梯度洗脫劑:A=1 ∶5~1 ∶1)分離得無色油狀液體103.63 g,產率75%;1H NMRδ: 7.26~7.24(m, 1H), 6.11(dd,J=6.2 Hz, 1.7Hz, 1H), 4.76~4.74(m, 1H), 3.72~3.70(m, 1H), 3.58(br, 1H), 1.69~1.65(m, 1H), 1.55(s, 9H), 0.95(d,J=2.5 Hz, 3H), 0.94(d,J=2.6 Hz, 3H);13C NMRδ: 168.9, 151.9, 148.5, 127.3, 84.1, 66.9, 31.2, 28.2, 20.4, 16.4; HR-ESI-MSm/z: Calcd for C13H21NO4Na+{[M+Na]+} 278.136 8, found 278.136 7。
在反應瓶中依次加入102.6 g(10 mmol)的CH2Cl2(100 mL)溶液,Et3N 4.2 mL(30 mmol), Ac2O 2.3 mL(20 mmol)和催化量DMAP(二甲氨基吡啶),攪拌下于室溫反應2 h。加入CH2Cl250 mL,靜置分層,有機相依次用飽和NaHCO3溶液和水洗滌,無水Na2SO4干燥,減壓蒸除溶劑后經硅膠柱層析(洗脫劑:A=1 ∶8)分離得無色晶體(E)-11 2.1 g,產率90%;1H NMRδ: 7.44(d,J=6.0 Hz, 1H), 6.51(d,J=10.2 Hz, 1H), 6.05(d,J=6.0 Hz, 1H), 2.81~2.76(m, 1H), 1.59(s, 9H), 1.12(d,J=6.6 Hz, 6H);13C NMRδ: 168.1, 149.8, 136.3, 134.8, 129.9, 122.6, 83.7, 28.2, 23.7; HR-ESI-MSm/z: Calcd for C13H19NONa+{[M+Na]+} 260.126 3, found 260.127 6。
在反應瓶中加入(E)-11 1.0 g(4.2 mmol)的CH2Cl2(40 mL)溶液,攪拌下于0 ℃慢慢加入TFA 13 mL,反應30 min。減壓除去過量的TFA,剩余物用乙酸乙酯(50 mL)溶解后依次經NaHCO3飽和溶液,水與飽和鹽水洗滌,無水Na2SO4干燥,減壓蒸除溶劑得無色油狀液體(E)-12 0.51 g,產率88%(無需純化,直接用于下一步反應)。
在反應瓶中加入(E)-12 0.51 g的苯(10 mL)溶液與催化量I2,攪拌下回流反應12 h(TLC跟蹤)。用亞硫酸鈉水溶液洗苯溶液至無色,再用水洗滌兩次,無水Na2SO4干燥,減壓除去溶劑后經硅膠柱層析(洗脫劑:A=1 ∶3)分離得無色油狀液體(Z)-10.51 g(quan)。
1.3 1的Baylis-Hillman反應
在反應瓶中加入(PhSe)2250 mg(0.8 mmol)的THF(2 mL)懸浮液,降溫至-15 ℃,氮氣保護,攪拌下緩慢滴加2.4 mol·L-1n-BuLi 0.34 mL(0.8 mmol),滴畢,制得無色苯硒基鋰溶液。降溫至-90 ℃,慢慢加入(E)-11 180 mg(0.5 mmol)和乙醛44 mg(1 mmol)的THF(2 mL)溶液,反應2 h。用飽和NH4Cl溶液淬滅反應,慢慢升至室溫,用正己烷(2×10 mL)萃取,合并有機相,用無水Na2SO4干燥,減壓蒸除溶劑后經硅膠柱層析(梯度洗脫劑:A=1 ∶50~1 ∶5)分離得無色油狀液體1377 mg,產率55%;1H NMRδ: 7.28(s, 1H), 6.56(d,J=10.5 Hz, 1H), 4.76(q,J=6.3 Hz, 1H), 2.88~2.77(m, 2H), 1.61(s, 9H), 1.49(d,J=6.6 Hz, 3H), 1.12(d,J=6.6 Hz, 6H);13C NMRδ: 167.8, 149.5, 137.9, 133.1, 130.4129.1, 83.8, 63.4, 28.2, 28.1, 23.6, 21.5; HR-ESI-MSm/z: Calcd for C15H23NO4Na+{[M+Na]+} 304.152 5, found 304.151 3。
在反應瓶中加入13 110 mg(0.39 mmol)的CH2Cl2(5 mL)溶液,攪拌下于0 ℃加入DMP 330 mg(0.80 mmol),于室溫反應3 h。加Et2O(10 mL)稀釋,過濾,濾液依次用半飽和的NaHCO3溶液與飽和食鹽水洗滌,無水Na2SO4干燥,減壓蒸除溶劑后經硅膠柱層析(洗脫劑:A=1 ∶10)分離得無色油狀液體14100 mg,產率90%;1H NMRδ: 8.13(s, 1H), 6.92(d,J=10.5 Hz, 1H), 3.01~2.83(m, 1H), 2.61(s, 3H), 1.64(s, 9H), 0.92(d,J=7.2 Hz, 6H);13C NMRδ: 193.1, 165.2, 149.7, 140.0, 137.6, 132.4, 130.6, 84.2, 29.8, 29.0, 28.2, 23.2。
2 結果與討論
2.1 1的合成
本文采用兩種不同的方法合成了1。
方法一:以鏈狀化合物為原料通過改良的HWE反應制備順式α,β-不飽和酯,最后經分子內親核取代反應進行環合(Scheme 1)。根據文獻[11]方法制備5:以Garner醛為原料先與異丙基溴格氏試劑反應制得3(d∶r>10 ∶1);用叔丁基二甲基硅基保護3的羥基,再脫掉丙叉基制得醇4;在TCCA/TEMPO條件下將醇4氧化成醛5。5經改良的HWE反應制備順式α,β-不飽和酯6;在TFA存在下脫掉6的氨基保護,進而通過分子內親核取代反應生成五元不飽和內酰胺7;在TFA存在下脫掉7的TBS保護基并進行消除反應合成了順式烯烴(Z-1)及其反式異構體E-1(Z∶E=1.5 ∶1.0),其構型經1H-1HCOSY譜進行確定,順式和反式產物通過硅膠柱層析分離。
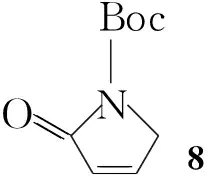
方法二:以環狀化合物為原料經Mukaiyama Aldol反應引入C5-位取代基(Scheme 1)。以吡咯為原料,經氧化及Boc保護氨基制得8; 8在TBSOTf 條件下形成烯醇硅醚化合物9[12];9在SnCl4催化下與異丁醛進行Mukaiyama aldol反應制得10(threo∶erythro=11∶1); 10(無論哪種構型)通過消除反應制得(E)-11 (經1H 1HCOSY譜確定其為反式構型); (E)-11 脫掉 Boc保護基制得反式烯烴(E)-12 ;加入碘,(E)-12 在苯中回流構型翻轉[13]制得(Z)-1。
2.2 (E)-11 的Baylis-Hillman反應
對于Myceliothermophin E的合成可以考慮采用 Baylis-Hillman反應將(Z)-1與2連接(Chart 1)。為了考察(Z)-1能否順利進行Baylis-Hillman反應,本文以(E)-11 和乙醛為原料,進行了Baylis-Hillman模型反應研究(Scheme 2),在(PhSe)2/n-BuLi條件下以較高產率(65%)合成了預期產物13;13經Dess-martin高碘喃(DMP)氧化順利的制得酮14。
[1] Yang Y L, Lu C P, Chen M Y,etal. Cytotoxic polyketides containing tetramic acid moieties isolated from the fungus myceliophthora thermophila:Elucidation of the relationship between cytotoxicity and stereoconfiguration[J].Chem Eur J,2007,13(24):6985-6991.
[2] Suzuki S, Hosoe T, Nozawa K,etal. Antifungal substances against pathogenic fungi,talaroconvolutins,from talaromyces convolutus[J].J Nat Prod,2000,63(6):768-772.
[3] Singh S B, Goetz M A, Jones E T,etal. Oteromycin:A novel antagonist of endothelin receptor[J].J Org Chem,1995,60(21):7040-7042.
[4] Osterhage C, Kaminksy R, K?nig G M,etal. Ascosalipyrrolidinone A,an antimicrobial alkaloid,from the obligate marine fungus ascochyta salicorniae[J].J Org Chem,2000,65(20):6412-6417.
[5] Lambert T H, Danishefsky S J. Total synthesis of UCS1025A[J].J Am Chem Soc,2006,128(2):426-427.
[6] Gelderblom W C A, Thiel P G, Marasas W F O,etal. Natural occurrence of fusarin C,a mutagen produced by Fusarium moniliforme,in corn[J].J Agric Food Chem,1984,32(5):1064-1067.
[7] Tan Z, Negishi E. Selective synthesis of epolactaene featuring efficient construction of methyl (Z)-2-iodo-2-butenoateand (2R,3S,4S)-2-trimethylsilyl-2,3-epoxy-4-methyl-g-butyrolactone[J].Org Lett,2006,8(13):2783-2785.
[8] Asami Y, Kakeya H, Onose R,etal. Azaspirene:A novel angiogenesis inhibitor containing a 1-oxa-7-azaspiro[4.4]non-2-ene-4,6-dione skeleton produced by the fungus neosartorya sp[J].Org Lett,2002,4(17):2845-2848.
[9] Iwasawa N, Maeyama K. A highly efficient synthesis of (-)-PI-091 construction of the 4-alkoxy-2-butene-4-lactam skeleton from fischer-type carbene complexes,alkynyllithiums,and tosyl isocyanate[J].J Org Chem,1997,62(7):1918-1919.
[10] Coleman R S, Walczak M C, Campbell E L. Total synthesis of lucilactaene,A cell cycle inhibitor active in p53-inactive cells[J].J Am Chem Soc,2005,127(46):16038-16039.
[11] Williams L, Zhang Z D, Shao F,etal. Grignard reactions to chiral oxazolidine aldehydes[J].Tetrahedron,1996,52(36):1673-11694.
[12] Curti C, Sartori A, Battistini L,etal. Vicarious silylative mukaiyama aldol reaction:A vinylogous extension[J].J Org Chem,2008,73(14):5446-5451.
[13] Rico R, Bermejo F. Stereoselective preparation of (5E)- and (5Z)-5-benzylidene-3-methyl-3-pyrrolin-2-ones.Application to the synthesis of ampullicine and isoampullicine[J].Tetrahedron Lett,1996,37(32):5809-5812.
猜你喜歡
中老年保健(2021年9期)2021-08-24 03:52:04
河北畫報(2021年2期)2021-05-25 02:07:46
中學生數理化(高中版.高考理化)(2020年2期)2020-04-21 05:33:04
兒童繪本(2020年5期)2020-04-07 17:46:30
兒童故事畫報(2019年5期)2019-05-26 14:26:14
Coco薇(2016年2期)2016-03-22 02:42:52
山東青年(2016年1期)2016-02-28 14:25:23
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年7期)2015-08-11 15:03:12
小雪花·成長指南(2015年4期)2015-05-19 14:47:56
