側鏈接有噁二唑的硅氧烷嵌段聚對苯乙烯撐衍生物的合成及其性能研究
2012-11-21 11:43:34吳軍寧羅新榮萬平玉
合成化學 2012年2期
關鍵詞:振動
吳軍寧, 羅新榮, 張 辰, 萬平玉
(北京化工大學 理學院,北京 100029)
有機電致發光材料是一種新型的光電材料,目前已經應用在通訊、信息和平面顯示等領域。在有機發光材料中研究最多,最被寄予厚望的是聚對苯乙烯撐(PPV)類聚合物發光材料[1]。但是純PPV由于含有苯環和乙烯的大共軛結構,導致分子產生淬滅現象,降低了熒光量子效率,同時也使PPV的熱穩定性差、不溶不熔,對其進行改性以達到應用的目的顯得尤為重要[2]。
本文以2,5-二甲基苯酚和4-氟苯甲酸乙酯為起始原料,經5步反應合成了單體2-(4-辛氧基苯基)-5-[4-(2,5-二溴甲基苯氧基)苯基]-1,3,4-噁二唑(5);以二甲基二氯硅烷和對甲酚為起始原料,經3步反應合成了單體二甲基二對溴甲基苯氧基硅烷(7)。在強堿條件下,5與7經Gilch反應成功合成了一種新型的側鏈接有噁二唑的硅氧烷嵌段聚對苯乙烯撐衍生物(8),其結構經FT-IR確證。用UV-Vis, FL和DSC研究了8的光學性能和熱穩定性。
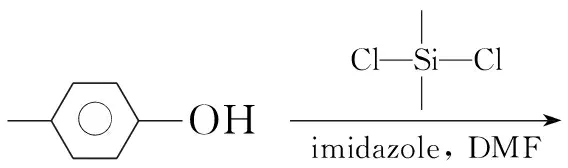
Scheme1
1 實驗部分
1.1 儀器與試劑
UV-3150型紫外-可見分光光度計;AV600型核磁共振儀(CDCl3為溶劑,TMS為內標);NIR型傅立葉紅外光譜儀(KBr壓片);F-7000型熒光分光光度計;Q100DSC型差示掃描量熱儀;1515型凝膠滲透色譜儀(GPC)。
叔丁醇鉀和對辛氧基苯甲酰氯參照文獻[7]方法制備;其余所用試劑均為分析純。
1.2 合成
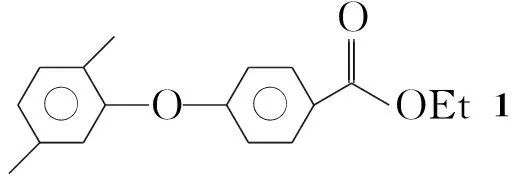
(1) 4-(2,5-二甲基苯氧基)苯甲酸乙酯(1)的合成
在反應瓶中加入2,5-二甲基苯酚12.58 g(103 mmol),叔丁醇鉀18.31 g(150 mmol)和DMF 80 mL,攪拌使其溶解;升溫至回流(150 ℃),緩慢滴加4-氟苯甲酸乙酯16.82 g(100 mmol)的DMF(30 mL)溶液,滴畢,回流反應16 h。冷卻至室溫,倒入過量去離子水中,分液,水層用無水乙醚(3×200 mL)萃取,合并萃取液,用無水硫酸鎂干燥,蒸除溶劑得棕色液體,經柱色譜[洗脫劑:V(正己烷) ∶V(二氯甲烷)=1 ∶1]純化得無色液體1 15.30 g,產率56%;1H NMRδ: 1.38~1.41(t, 3H, CH3), 2.15~2.33(s, 6H, ArCH3), 6.82, 6.88~6.90, 6.97~6.98, 7.16~7.18, 7.99~8.01(m, 7H, ArH)。
(2) 4-(2,5-二甲基苯氧基)苯甲酰肼(2)的合成[3]
在反應瓶中加入80%水合肼23.00 g(460 mmol)和無水乙醇30 mL,攪拌下升溫至回流(90 ℃),緩慢滴加1 12.48 g(46 mmol)的乙醇(20 mL)溶液,滴畢,回流反應30 h。冷卻至室溫,除去溶劑后用無水甲醇重結晶得白色晶體2 4.3 g,產率36.5%, m.p.112 ℃~114 ℃;1H NMRδ: 2.13, 2.30(s, 6H, ArCH3), 4.0(b, 2H, NH2), 7.24(b, 1H, NH), 6.78, 6.88~6.90, 6.94~6.95, 7.68~7.69(m, 7H, ArH)。
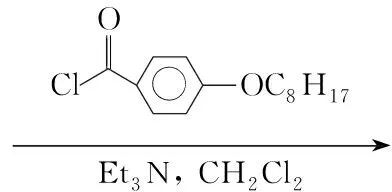
(3)N-(4-辛氧基苯甲酰基)-4-(2,5-二甲基苯氧基)苯甲酰肼(3)的合成
在反應瓶中加入24.0 g(15.6 mmol),三乙胺1.58 g(15.6 mmol)和二氯甲烷50 mL,攪拌下于室溫緩慢滴加對辛氧基苯甲酰氯4.19 g(15.6 mmol),滴畢,于室溫反應6 h。倒入去離子水中,分液,水層用二氯甲烷(3×100 mL)萃取,合并萃取液,用無水硫酸鎂干燥,蒸除溶劑后用甲醇重結晶得白色晶體3 6.36 g,產率86.6%, m.p.153 ℃~156 ℃;1H NMRδ: 0.89~0.93(t, 3H, CH3),1.31~1.85(m, 12H, CH2), 2.14, 2.32(s, 6H, ArCH3), 3.98~4.01(t, 2H, OCH2), 6.78~6.97, 7.15~7.17, 7.82~7.84(m, 11H, ArH), 9.55~9.66(d, 2H, NH)。
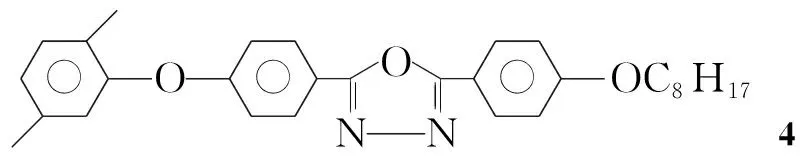
(4) 2-(4-辛氧基苯基)-5-[4-(2,5-二甲基苯氧基)苯基]-1,3,4-噁二唑(4)的合成[4]
在反應瓶中加入3 6.35 g(13 mmol),二氯亞砜5.89 g(50 mmol)及苯80 mL,攪拌下回流(90 ℃)反應6 h。冷卻至室溫,倒入過量去離子水中,分液,水層用氯仿(3×150 mL)萃取,合并萃取液,用無水硫酸鎂干燥,蒸出溶劑后經硅膠柱色譜[洗脫劑:A=V(正己烷) ∶V(乙酸乙酯)=15 ∶1]純化得白色晶體4 5.03 g,產率82.3%, m.p.76 ℃~78 ℃;1H NMRδ: 0.88~0.90(t, 3H, CH3), 1.30~1.83(m, 12H, CH2), 2.16, 2.32(s, 6H, ArCH3), 4.03~4.05(t, 2H, OCH2), 6.83, 6.96~7.03, 7.16~7.18, 8.05~8.07(m, 11H, ArH)。
(5) 5的合成[5]
在反應瓶中加入45.00 g(10.6 mmol),N-溴代丁二酰亞胺(NBS)3.91 g(22 mmol),過氧化苯甲酰(BPO)30 mg和四氯化碳100 mL,攪拌下回流(90 ℃)反應至終點(TLC檢測)。過濾,濾液倒入過量去離子水中,分液,水層用二氯甲烷(3×150 mL)萃取,合并萃取液,用無水硫酸鎂干燥,蒸出溶劑后經硅膠柱色譜(洗脫劑:A=20 ∶1)純化得白色晶體5 1.83 g,產率27.6%, m.p.113 ℃~115 ℃;1H NMRδ: 0.88~0.90(t, 3H, CH3), 1.30~1.82(m, 12H, CH2), 4.02~4.05(t, 2H, OCH2), 4.40, 4.55(s, 4H, CH2Br), 6.98~7.02, 7.14~7.21, 8.04~8.13(m, 11H, ArH);13C NMRδ: 14.0, 22.6, 26.0, 27.0, 29.1, 29.2, 29.3, 31.7, 32.0, 68.3, 115.0, 116.1, 118.8, 119.5, 120.1, 125.2, 128.6, 129.7, 131.9, 140.4, 154.2, 159.6, 162.0, 163.6, 164.5。
(6)二甲基二對甲基苯氧基硅烷(6)的合成[6]
在反應瓶中依次加入對甲基苯酚11.56 g(107 mmol),咪唑7.28 g(107 mmol)及DMF 45 mL,攪拌下于室溫緩慢滴加二甲基二氯硅烷6.45 g(50 mmol),滴畢,N2保護下于22 ℃反應36 h。倒入去離子水中,分液,水層用二氯甲烷(3×200 mL)萃取,合并萃取液,用無水硫酸鈉干燥,蒸出溶劑后經硅膠柱色譜(洗脫劑:正己烷)純化得無色液體6 9.20 g,產率67.65%;1H NMRδ: 0.36(s, 6H, CH3), 2.29(s, 6H, ArCH3), 6.84~6.86(d, 4H, ArH), 7.04~7.06(d, 4H, ArH);29Si NMRδ: -5.62(s, Si)。
(7)7的合成[6]
在反應瓶中加入62.46 g(9.0 mmol), NBS 3.56 g(20 mmol), BPO 20 mg及四氯化碳25 mL,攪拌使其溶解;回流(85 ℃)反應至終點(TLC檢測)。過濾,濾液蒸出溶劑后用水洗滌,水液用二氯甲烷萃取,合并萃取液,用無水硫酸鈉干燥,減壓蒸除溶劑后用甲苯稀釋得7的甲苯溶液。1H NMRδ: 0.40(s, 6H, CH3), 4.48(s, 4H, CH2), 6.90~6.91(d, 4H, ArH), 7.26~7.29(d, 4H, ArH)。
(8)8的合成
N2保護。在反應瓶中加入50.50 g(0.8 mmol)和無水甲苯40 mL,快速攪拌下加入70.43 g(0.1 mmol)的甲苯溶液,攪拌10 min。緩慢滴加1 mol·L-1叔丁醇的無水THF溶液2.7 mL,滴畢,于室溫反應10 h(溶液由無色變為黃色)。加入少量5的單溴代化合物封端,繼續反應1 h。倒入400 mL甲醇中,離心分離得黃色固體,用水洗滌后經索氏提取器(溶劑甲醇)除去小分子聚合物,再用甲醇(300 mL)分級沉淀,抽濾,濾餅干燥得黃色固體8 80 mg,產率10%。
2 結果與討論
2.1 表征
8的IR譜圖見圖1。由圖1可見,3 030 cm-1為=C-H伸縮振動峰,1 640 cm-1為C=C伸縮振動峰,峰強度較弱是因為C=C位于鏈狀PPV的中心,對稱性很強。1 010 cm-1為亞乙烯基反式H的面外彎曲振動峰,推斷形成C=C反式結構。1 611 cm-1, 1 510 cm-1和1 491 cm-1為苯環C-H伸縮振動峰,836 cm-1為苯環C-H的面外振動,證明含有苯環。2 925 cm-1和2 855 cm-1為甲基和亞甲基的反對稱和對稱C-H伸縮振動峰,1 419 cm-1和1 375 cm-1分別為甲基反對稱彎曲振動和對稱彎曲振動峰;744 cm-1為亞甲基面內搖擺振動峰,證明含有甲基和亞甲基結構。1 248 cm-1為Ph-O-C反對稱伸縮振動峰(與Si-OPh伸縮振動重合),1 110 cm-1為Ph-O-C對稱伸縮,證明含有芳香醚結構。1 068 cm-1為C=N反對稱伸縮峰。1 248 cm-1和1 169 cm-1為Si-OPh反對稱和對稱伸縮峰,873 cm-1為O-SiMe特征峰,證明硅氧烷已經嵌段到PPV中。
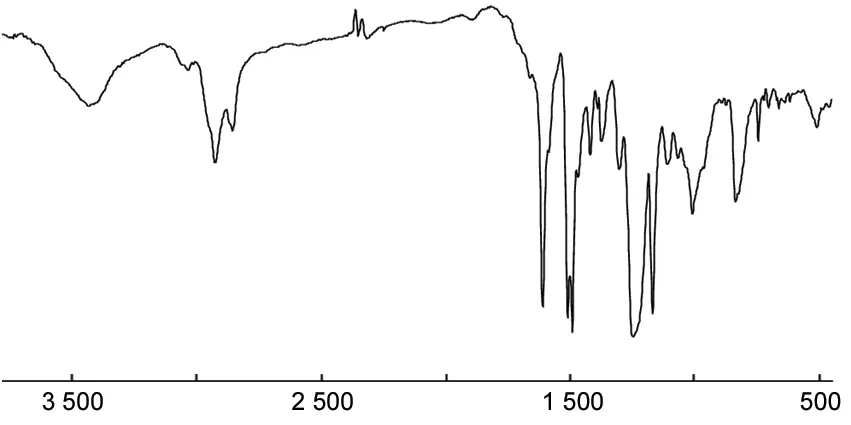
ν/cm-1圖1 8的IR譜圖Figure 1 IR spectrum of 8
2.2 光學性能
(1) UV-Vis
8的UV-Vis譜圖見圖2。由圖2可見,8有兩個最大吸收峰,302 nm對應側鏈噁二唑基團的吸收,410 nm對應PPV中苯環、乙烯單元的π-π*電子躍遷。8分子中噁二唑基團和PPV單元都保持了各自的吸收,PPV對應的吸收峰與噁二唑吸收峰相比很小,這是因為硅氧基團嵌段了PPV的共軛主鏈,影響到其紫外吸收的強度。
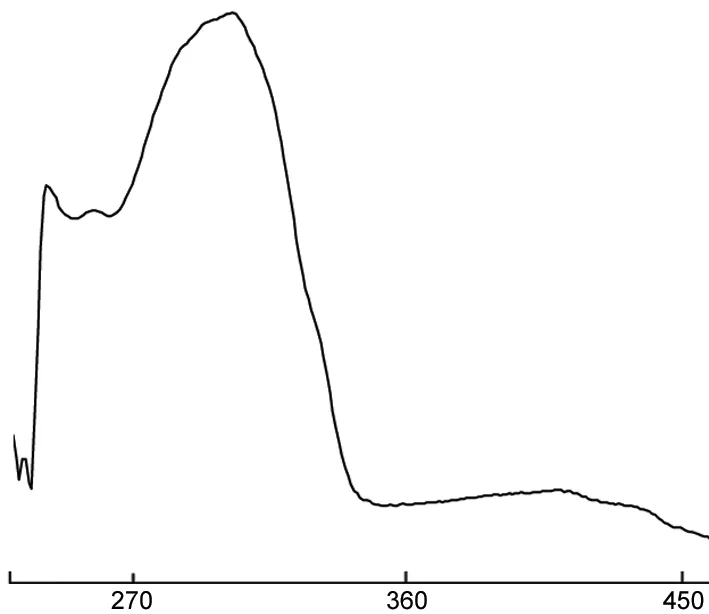
λ/nm圖2 8的UV-Vis譜圖Figure 2 UV-Vis spectrum of 8
(2) 熒光光譜
8的熒光光譜圖見圖3。由圖3可見,8的最大發射峰出現在480 nm,屬于藍光的波長范圍內,為藍色有機電致發光材料。
2.3 熱穩定性
8的DSC曲線見圖4。由圖4可見,8的玻璃化溫度為201.112 ℃,說明將O-Si-O基團嵌段的改性PPV在熱穩定性方面有了極大的改善。
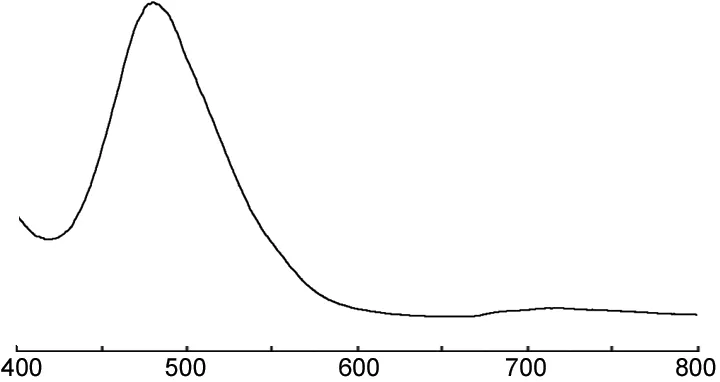
λ/nm圖3 8的熒光光譜圖*Figure 3 Fluorescence spectrum of 8*激發波長384 nm,氯仿為溶劑,c=0.1 mmol·L-1
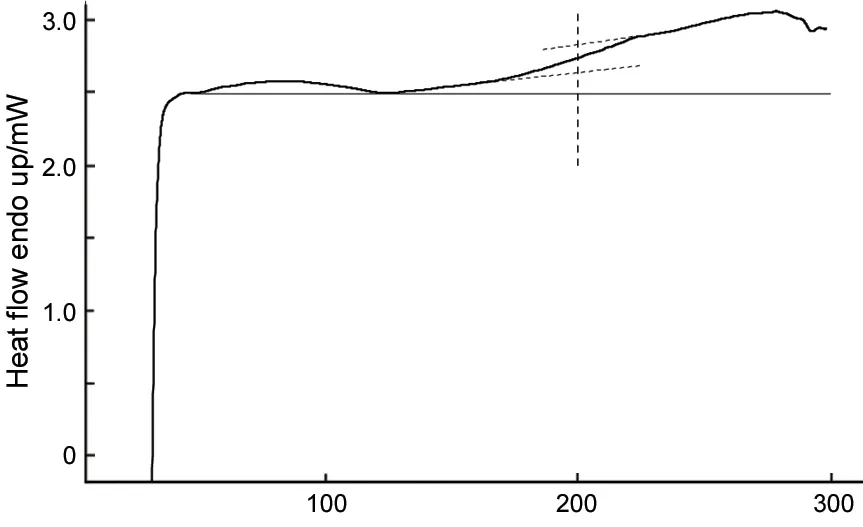
Temperature/℃圖4 8的DSC曲線Figure 4 DSC curve of 8
我們還測定了8的分子量,其Mn=112 825, Mw=116 387,說明聚合程度較大,8為高分子量的聚合物。
3 結論
以2,5-二甲基苯酚,4-氟苯甲酸乙酯,二甲基二氯硅烷和對甲酚為原料,通過8步反應合成了PPV改性材料,在PPV側鏈引入電子傳輸基團噁二唑,主鏈引入O-Si-O的結構嵌段大共軛體系,從而避免了熒光淬滅的問題,也改善了其熱穩定性并且實現了藍色發光。
[1] Burroughes J H, Bradley Brown A R, Marks R N,etal. Light-emitting diodes based on conjugated polymers[J].Nature,1990,347:539-541.
[2] Andrew C Grimsdale, Khai Leok Chan, Rainer E Martin,etal. Synthesis of light-emitting conjugated polymers for applications in electroluminescent devices[J].Chemical Reviews,2009,109(3):897-1091.
[3] Joo Hyun Kima, Hoosung Lee. Synthesis and characterization of efficient orange-red emitting poly(p-phenylenevinylene) derivatives with 1,3,4-oxadiazole unit[J].Synthetic Metals,2007,157:1040-1045.
[4] Haoyu Tang, Naiheng Song. Synthesis and properties of 1,3,4-oxadiazole-containing high-performance bismaleimide resins[J].Polymer,2007,48:129-138.
[5] Hailin Li, Sishun Kang, Zhitao Xing. The synthesis,optical properties and X-ray crystal structure of novel 1,3,4-oxadiazole derivatives carrying a thiophene unit[J].Dyes and Pigments,2009,80:163-167.
[6] Piyush Kumar, Weizhong Zheng, Stephen A McQuarrie,etal.18F-FESB:Synthesis and automated radiofluorination of a novel18F-labeled pet tracer for b-amyloid plaques[J].J Label Compd Radiopharm,2005,48:983-996.
[7] Thayer F K. Acethylmandelic Acid and Acetylmandylchloride[M].Organic Synthesis,1947,2:12-13.
猜你喜歡
科學大眾(2023年17期)2023-10-26 07:39:14
大電機技術(2022年5期)2022-11-17 08:12:48
天天愛科學(2020年6期)2020-09-10 07:22:44
瘋狂英語·新讀寫(2020年3期)2020-06-06 09:05:56
數學物理學報(2018年4期)2018-09-14 03:40:58
數學物理學報(2017年6期)2018-01-22 02:26:40
船海工程(2015年4期)2016-01-05 15:53:26
噪聲與振動控制(2015年4期)2015-01-01 07:08:44
計算物理(2014年2期)2014-03-11 17:01:44
鄭州大學學報(理學版)(2014年3期)2014-03-01 04:21:00
