非鉻系乙烯三聚催化劑的研究進展
2012-09-08 07:34:26隋軍龍
合成樹脂及塑料 2012年5期
隋軍龍
(中國石油化工股份有限公司北京北化院燕山分院,北京市102500)
非鉻系乙烯三聚催化劑的研究進展
隋軍龍
(中國石油化工股份有限公司北京北化院燕山分院,北京市102500)
根據(jù)催化劑活性中心金屬元素的不同,綜述了鋯系、釩系、鉭系、鈦系和鎳系五類非鉻系乙烯三聚催化劑的研究情況,介紹了催化劑的結構特征、催化乙烯三聚的反應機理、反應特點等。比較了配體結構、反應溫度、反應壓力、助催化劑用量等因素對催化劑活性、1-己烯選擇性的影響。其中,尤以鈦系催化劑最為突出,不僅活性高,對1-己烯的選擇性也高,但甲基鋁氧烷的用量大是該體系的一個弱點。
乙烯 三聚 非鉻系催化劑 反應機理
1-己烯是生產(chǎn)線型低密度聚乙烯的主要共聚單體和合成高級增塑劑的重要原料。自從1996年Philips公司采用鉻系催化劑[1]首次實現(xiàn)由乙烯選擇性三聚制1-己烯的工業(yè)化以來,對乙烯三聚催化劑的開發(fā)研究一直都是聚烯烴領域內(nèi)的熱點,尤其以鉻系催化劑最為突出,配體涵蓋了芳香族、雜原子和多齒配體等[2-8]。從對催化劑活性與選擇性的改進、鉻對環(huán)境的影響等角度考慮,很多學者研究了非鉻系配合物作為乙烯三聚催化劑的活性組分(如鋯、釩、鉭和鈦等前過渡金屬配合物以及鎳等后過渡金屬配合物)。本文綜述了這幾類非鉻系催化劑在乙烯三聚方面的研究進展,以期促進我國乙烯三聚非鉻系催化劑體系的基礎和應用研究。
1 鋯系催化劑
菲利普斯石油公司用乙酰丙酮鋯[Zr(acac)2]與吡咯(Pyrrole)、三乙基鋁(TEAL)組成的三元催化劑體系催化乙烯三聚制備1-己烯[9]。以環(huán)己烷為溶劑,在乙烯壓力為3.8 MPa,反應溫度為80℃、n[Zr(acac)2]/n(Pyrrole)/n(TEAL)=1 ∶3 ∶22 的條件下,制得質(zhì)量分數(shù)為26.8%的液體產(chǎn)物和73.2% 的聚乙烯(PE)。液體產(chǎn)物中w(C6)為26.9%,1-己烯的選擇性為100.0%,催化劑的活性很低,僅有29 g/(g·h)。
Wang Mei等[10]研究發(fā)現(xiàn),二茂鋯化合物能夠催化乙烯三聚得到亞甲基環(huán)戊烷(MCP)。作者認為這種環(huán)狀的C6異構體是由于反應中形成的鋯五元環(huán)中間體消除β-H得到的。實驗結果表明:TEAL 為助催化劑,在 n(Al)/n(Zr)=100 ∶1,150 ℃,1.4 MPa下,MCP的選擇性最高,達43.0%。相同條件下,以乙基鋁氧烷(EAO)為助催化劑,MCP的選擇性為27.0%。進一步降低EAO的用量,當n(Al)/n(Zr)=25 ∶1 時,1-己烯的選擇性為55.0%,MCP的選擇性僅為3.0%,這時催化劑的活性僅為153 g/(g·h)。 主要副產(chǎn)物是 PE和少量的1,5-己二烯。
Sevn等[11]用梯度校正密度泛函方法對硼雜苯-芳烴鋯配合物的乙烯三聚性能進行了理論研究。經(jīng)計算,他們認為C1橋連的鋯配合物有可能是高效的乙烯三聚催化劑,但目前還未見有此類文章報道。
綜上所述,目前乙烯三聚鋯系催化劑的活性都較低,生成1-己烯的選擇性為55.0%左右,而且會生成大量的PE,這些都增加了鋯系催化劑工業(yè)化的不確定性。
2 釩系催化劑
菲利普斯石油公司用乙酰丙酮氧釩[VO(acac)2]與Pyrrole,TEAL組成三元催化劑體系催化乙烯三聚制1-己烯[9]。以環(huán)己烷為溶劑,在乙烯壓力為3.8 MPa,反應溫度80 ℃,n[VO(acac)2]/n(Pyrrole)/n(TEAL)=1 ∶3 ∶22時,制得質(zhì)量分數(shù)為56.8% 的液體產(chǎn)物和43.2% 的PE。液體產(chǎn)物中w(C6)為63.0%,1-己烯的選擇性為79.4%,但催化劑的活性仍不高,為115 g/(g·h)。

Santi等[12]制備的催化劑是低氧化態(tài)的釩化合物,通式為 R2′VX。 其中,R′可以是苯或單、雙、三烷基取代的苯,X 可以是 Cl,Br,I,B(Ar)4-,AlCl4-,羧化物,磺酸鹽。實驗結果表明:加入少量某些雜環(huán)化合物(與釩的摩爾比為1∶20),如Pyrrole、吡唑、菲咯啉、嘧啶等能引發(fā)反應,甚至不用助催化劑。在室溫,乙烯壓力為0.7 MPa,V(Ph)2(Ph 為苯基,下同)與 Cp2Fe(BPh4)(Cp 為茂環(huán),下同)以1 ∶1的摩爾比混合就能引發(fā)乙烯三聚,催化劑活性達333 g/(g·h), 反應混合物中含質(zhì)量分數(shù)為99.0%的1-己烯。
盡管釩系催化劑催化乙烯三聚可以不用助催化劑,但活性太低仍是制約它不能工業(yè)化的主要因素。
3 鉭系催化劑
Murtuza等[13]在以鹵化烷基鋁為助催化劑,用TaCl5在氯苯或己烷中催化乙烯聚合時檢測到高度支化的PE油品,由此發(fā)現(xiàn)了鉭也能作為乙烯三聚的催化劑。支化油品的形成只能用雙催化機理來解釋:烷基鉭先催化乙烯齊聚得到α-烯烴,α-烯烴再進一步發(fā)生陽離子齊聚。催化體系中還含有2,6-二叔丁基吡啶,它是陽離子齊聚的抑制劑。 在45 ℃,4.8 MPa,氯苯為溶劑,TaCl5、二氯乙基鋁、2,6-二叔丁基吡啶的摩爾比為1∶1∶1,反應持續(xù)16 h以上,可得到含質(zhì)量分數(shù)為45.0%的1-己烯和55.0%具有Schulz-Flory分布(分布系數(shù)為0.73)的α-烯烴,這證明了該體系的雙催化機理。實驗還發(fā)現(xiàn),TaCl5在三烷基鋁、四烷基氯化銨的共同作用下,在氯苯中催化乙烯齊聚能提高三聚體的選擇性:質(zhì)量分數(shù)為90.0%的液體產(chǎn)品中含質(zhì)量分數(shù)為75.0%的1-己烯。
Andes等[14]開發(fā)出一種選擇性極高的三聚催化劑體系,該體系中除TaCl5外,還含有與TaCl5等摩爾比的甲基化試劑(如二甲基鋅)。在45℃,4.8 MPa,氯苯做溶劑的條件下,1-己烯的總選擇性達到96.0%,催化劑活性為214 g/(g·h)。
核磁共振氫譜分析表明,TaCl5發(fā)生烷基化形成的Ta(CH3)2Cl3是活性催化劑的前驅(qū)體。與其他體系不同的是,該催化劑體系沒有配體,有效的三聚物種Ta(Ⅲ)Cl3被認為是“裸露的”,然而極有可能的是氯苯溶劑在催化過程中扮演配位的角色。
4 鈦系催化劑
菲利普斯石油公司用乙酰丙酮氧鈦[TiO(acac)2]與Pyrrole,TEAL組成三元催化體系進行乙烯三聚[9]。以環(huán)己烷為溶劑,乙烯壓力為3.8 MPa,反應溫度為80 ℃,n[TiO(acac)2]/n(Pyrrole)/n(TEAL)=1∶3∶22的條件下,制得質(zhì)量分數(shù)為71.2% 的液體齊聚產(chǎn)物。其中,w(C6)為55.0%,1-己烯的選擇性為87.3%,催化劑活性為222 g/(g·h)。
鈦系三聚催化劑是Pellecchia等用Cp*TiMe3-B(C6F5)3(Me 為甲基,Cp* 為五甲基茂基,下同)體系催化劑生產(chǎn)支化的線型低密度聚乙烯時發(fā)現(xiàn)的[15]。聚合物只有丁基支鏈,而通過氣相色譜和核磁共振氫譜分析,在液相組分中檢測出高選擇性的1-己烯。該催化劑的三聚和共聚合能力同樣表現(xiàn)在乙烯與苯乙烯的共聚合中[16]。乙烯和苯乙烯反應生成的6-苯基-1-己烯與乙烯共聚合生成具有4-苯基-1-丁基支鏈的PE。其他種類的鈦前驅(qū)體[如 Cp*TiCl3,CpTiCl3或(Ind)TiCl3(Ind 為茚基)]與甲基鋁氧烷(MAO)作用則會抑制共聚合,生成質(zhì)量分數(shù)高達90.0%的苯基己烯低聚物。
Zhu Fangming 等[17]發(fā)現(xiàn),(Cp*)Ti(OCH2Ph)3和改性MAO組成的催化劑體系會催化生成有乙基和丁基支鏈的聚合物。他們認為這是由于反應開始生成的1-丁烯和1-己烯與乙烯共聚合形成的,而且他們還提出用反應過程中鈦五元環(huán)和鈦七元環(huán)中間體的形成機理來解釋選擇性二聚體和三聚體的生成。
Deckers等[18-22]開發(fā)出了高活性和高選擇性的非鉻基乙烯三聚催化劑。在Cp*TiMe3-B(C6F5)3體系中甲苯溶劑能夠穩(wěn)定活性Ti(Ⅱ)三聚催化劑物種[15],這個發(fā)現(xiàn)使他們找到了新型催化劑設計的突破點。他們在茂環(huán)上引入一個芳基來模仿溶劑所起的穩(wěn)定角色,實驗結果表明,引入的芳基在使催化劑由聚合轉變?yōu)槿蹠r起了至關重要的作用。這種芳烴側基可以看作是一種半穩(wěn)定配體,既可以起穩(wěn)定金屬活性中心的作用,同時也能使單體很容易接近活性中心。
Deckers等[18]用[CpCMe2Ph]TiCl3作催化劑的前驅(qū)體,MAO 為助催化劑,在 n([CpCMe2Ph]TiCl3)/n(MAO)=1 ∶1000,反應溫度為30 ℃,乙烯壓力為0.2 MPa下催化乙烯三聚得到含質(zhì)量分數(shù)為98.4%的液體齊聚物和1.6%的PE。其中,液體齊聚物中的 w(C6)為87.0%(1-己烯的選擇性為98.0%),w(C10)為11.0%,w(C12)為0.4%。 催化劑活性為22354 g/(g·h)。[η5-C5H4CMe2Ph]TiMe3(η5指配體與金屬有5個配位點)作為前驅(qū)體與[PhMe2NH][B(C6F5)4],B(C6F5)3或 MAO/SiO2一起作用也是高效的乙烯三聚催化劑。
Deckers等考察了幾種參數(shù)對催化劑性能的影響,包括:1)半穩(wěn)定芳烴側基;2)芳香族溶劑和脂肪族溶劑的對比;3)在茂環(huán)和芳環(huán)之間的橋基;4)芳環(huán)上的取代基;5)茂環(huán)上的取代基。結果表明:含CMe2橋基、茂環(huán)上具有SiMe3取代基、芳環(huán)上具有烷基取代基的催化劑體系催化乙烯三聚的活性最高。這三個主要參數(shù)之間空間位阻和電子效應的相互作用對催化劑的活性和選擇性影響較大。
在 Deckers等研究的基礎上,Huang Jiling等[23-24]用噻吩基代替芳烴側基制得配合物,用摩爾質(zhì)量是該配合物1000倍的MAO活化,于30℃,乙烯壓力為0.8 MPa進行實驗評價。結果表明:噻吩基通過碳橋連接茂環(huán)的配合物能夠催化得到1-己烯,1-己烯的總選擇性為86.0%,催化劑活性為4596 g/(g·h)。 這是因為 η1-S 對 Ti中心的配位有助于初期Ti(Ⅱ)活性物種的穩(wěn)定。
Arno等[25]通過理論計算進一步支持了Deckers等提出的鈦系乙烯三聚過程中金屬環(huán)的機理(見圖1)。但有一點與Deckers不同的是:Arno認為從鈦七元環(huán)中間體到(1-己烯)鈦配合物的形成是由于氫直接從β碳轉移到α碳上實現(xiàn)的,而不是通過β-H消除/還原實現(xiàn)的(見圖2)。
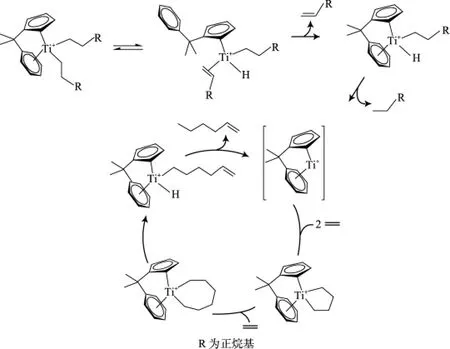
圖1 Deckers等提出的乙烯三聚催化機理[19]Fig.1 Catalytic mechanism of ethylene trimerization proposed by Deckers,et al
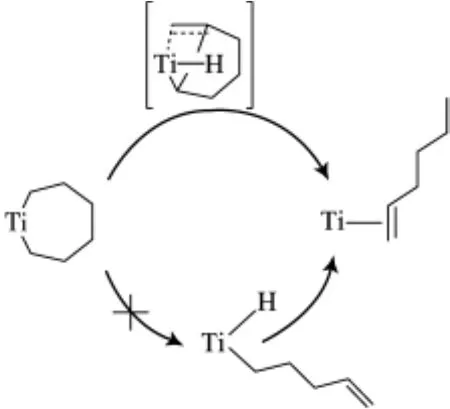
圖2 Arno等提出的1-己烯鈦配合物的形成機理Fig.2 Formation mechanism of the1-hexene titanium complex proposed by Arno,et al
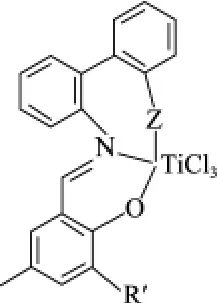
圖3 具有供電子基團修飾的苯氧基-亞胺鈦配合物Fig.3 Phenoxy-imine titanium complexes with pendant electron donors
日本三井化學公司在其研究的烯烴聚合催化劑(又稱FI催化劑)的基礎上,對苯氧基-亞胺配體用供電子基團芳基 —OMe進行修飾,得到了高活性的鈦系乙烯三聚催化劑[26](見圖3)。
以MAO為助催化劑、環(huán)己烷為溶劑。配合物1在乙烯壓力為0.8 MPa,反應溫度為80 ℃,n(Al)/n(Ti)=10000 ∶1 的條件下,催化得到產(chǎn)物中w(C6)為76.6%,w(PE)為23.4%,催化劑活性為5 kg/(g·h)。配合物2在乙烯壓力為0.8 MPa,反應溫度80℃,n(Al)/n(Ti)=10000 ∶1 的條件下,催化劑活性上升到119 kg/(g·h),產(chǎn)物包括 C6,C10,PE。 其中,C6的選擇性上升到86.2%,1-己烯的產(chǎn)率為138 kg/(g·h)。作者認為是 —OMe在 Ti(Ⅱ)活性物種的形成和穩(wěn)定中起到了至關重要的作用。配合物3在乙烯壓力為0.8 MPa、反應溫度為80 ℃、n(Al)/n(Ti)=10000 ∶1的條件下,催化劑活性升至169 kg/(g·h),C6的選擇性上升到91.4%,1-己烯的產(chǎn)率為155 kg/(g·h)。 將乙烯壓力增至5.0 MPa,其他條件不變,配合物3的催化活性猛增到7140 kg/(g·h),C6的選擇性上升到92.3%,1-己烯的產(chǎn)率為6590 kg/(g·h)。這個活性是迄今為止乙烯三聚催化劑報道的最高活性,比商業(yè)化的鉻系催化劑活性要高得多。
總的來說,盡管從活性和選擇性上,鈦系三聚催化劑能夠與鉻系競爭,但鈦系體系中MAO的大量使用卻是它最大的缺陷。
5 鎳系催化劑
大多數(shù)乙烯三聚催化劑都基于前過渡金屬(如 Cr,Ti,V,Ta),然而某些后過渡金屬(如 Ni)也可以形成三聚催化劑。
菲利普斯石油公司用乙酰丙酮鎳[Ni(acac)2]與Pyrrole,TEAL組成三元催化劑體系進行乙烯三聚[9]。以環(huán)己烷為溶劑,在乙烯壓力為3.8 MPa,溫度為80 ℃,n[Ni(acac)2]/n(Pyrrole)/n(TEAL)=1∶3∶22的條件下,催化得到質(zhì)量分數(shù)為95.4%的液體三聚物,其中,w(C6)為69.0%,1-己烯的選擇性為65.0%,催化劑活性為324 g/(g·h)。
日本三井化學公司用NiBr2(PhN=CHC5H4N)與 MAO組成二元催化體系進行乙烯三聚[27]。在乙烯壓力為0.1 MPa,反應溫度為25℃,n[NiBr2(PhN=CHC5H4N)]/n(MAO)=1 ∶250的條件下,催化得到的液體產(chǎn)物中含質(zhì)量分數(shù)為60.0%的三聚物,24.0%的二聚物和16.0%的四聚物,催化劑活性為10 kg/(g·h)。
(Ph3P)2NiBr2也能被 MAO[28]或 MeAlCl2[29]活化形成三聚催化劑。在乙烯壓力為3.0 MPa,反應溫度為100 ℃,n[(Ph3P)2NiBr2]/n(MeAlCl2)=1 ∶300 的條件下,催化得到的液體產(chǎn)物中含有質(zhì)量分數(shù)為95.0% 的三聚物,催化劑活性為118 kg/(g·h)。 其中,三聚物中含有質(zhì)量分數(shù)為47.0%的3-甲基-2-戊烯產(chǎn)品、21.0% 的2-乙基-1-丁烯產(chǎn)品和25.0%的2-己烯產(chǎn)品。
6 結語
近年來,從對乙烯三聚催化劑活性與選擇性的改進、鉻對環(huán)境的影響等角度考慮,不斷有關于非鉻系乙烯三聚催化劑的報道,其中,尤以鈦系催化劑最為突出,不僅活性高,對1-己烯的選擇性也高,但MAO的用量大是該體系的一個弱點。因此,如何兼顧高活性和高選擇性的同時,降低MAO的用量或者使用價格低廉的烷基鋁作助催化劑是當前研究的重點。
[1] Freeman J W,Buster J L,Knudsen R D.Olefin production:US,5856257[P].1999-01-05.
[2] David S McGuinness,Peter Wasserscheid,Wilhelm Keim,et al.First Cr(Ⅲ)-SNS complexes and their use as highly efficient catalysts for the trimerization of ethylene to1-hexene[J].J Am Chem Soc,2003,125(18):5272-5273.
[3] Amir Jabri,Claire Temple,Patrick Crewdson,et al.Role of the metal oxidation state in the SNS-Cr catalyst for ethylene trimerization:isolation of di-and trivalent cationic intermediates[J].J Am Chem Soc,2006,128(28):9238-9247.
[4] Claire N Temple,Sandro Gambarotta,Ilia Korobkov,et al.New insight into the role of the metal oxidation state in controlling the selectivity of the Cr-(SNS)ethylene trimerization catalyst[J].Organometallics,2007,26(18):4598-4603.
[5] Khalid Albahily,Danya Al-Baldawi,Sandro Gambarotta,et al.Preparation and characterization of a switchable single-component chromium trimerization catalyst[J].Organometa llics,2008,27(21):5708-5711.
[6] Igor Y Skobelev,Valentina N Panchenko,Oleg Y Lyakin,et al. In situ EPR monitoring of chromium species formed during Cr-pyrrolyl ethylene trimerization catalyst formation[J].Organometallics,2010,29(13):2943-2950.
[7] Sebastiano Licciulli,Khalid Albahily,Valeria Fomitcheva,et al.A chromium ethylidene complex as a potent catalyst for selective ethylene trimerization[J].Angew Chem Int Ed,2011,50(10):2346-2349.
[8] Khalid Albahily,Yacoob Shaikh,Zeeshan Ahmed,et al.Isolation of a self-activating ethylene trimerization catalyst of a Cr-SNS system[J].Organometallics,2011,30(15):4159-4164.
[9] Reagan W K,Freeman J W,Conroy B K,et al.Process for olefin polymerization:US,5451645[P].1995-09-19.
[10] Wang Mei,Shen Yumei,Qian Mingxing,et al.Oligomerization and simultaneous cyclization of ethylene to methylenecyclopentane catalyzed by zirconocene complexes[J].J Organomet Chem,2000,599(2):143-146.
[11] Sven Tobisch,Tom Ziegler.Catalytic oligomerization of ethylene to higher linear α-olefins promoted by cationic group4 cyclopentadienyl-arene active catalysts:toward the computational design of zirconium-and hafnium-based ethylene trimerization catalysts[J].Organometallics,2005,24(2):256-265.
[12] Santi R,Romano A M,Grande M,et al.Process for the preparation of1-hexene:WO,01/68572[P].2001-09-20.
[13] Murtuza S,Harkins S B,Long G S,et al.Tantalum-and titanium-based catalytic systems for the synthesis of hyperbranched polyethene[J].J Am Chem Soc,2000,122(9):1867-1872.
[14] Andes C,Harkins S B,Murtuza S,et al.New tantalumbased catalyst system for the selective trimerization of ethene to1-hexene[J].J Am Chem Soc,2001,123(30):7423-7424.
[15] Pellecchia C,Pappalardo D,Gruter G.Branched polyethylene produced by a half-titanocene catalyst[J].Macromolecules,1999,32(13):4491-4493.
[16] Pellecchia C,Pappalardo D,Oliva L,et al.Selective cooligomerization of ethylene and styrenes by half-titanocene catalysts and synthesis of polyethylenes with4-Aryl-1-butyl branches[J].Macromolecules,2000,33(8):2807-2814.
[17] Zhu Fangming,Huang Yun,Yang Yijia,et al.Branched polyethylene prepared by in situ copolymerization of ethylene with monotitanocene and modified methylaluminoxane catalyst[J].J Polym Sci A,2000,38(23):4258-4263.
[18] Deckers P J W,Hessen B,Teuben J H.Catalytic trimerization of ethene with highly active cyclopentadienyl-arene titanium catalysts[J].Organometallics,2002,21(23):5122-5135.
[19] Deckers P J W,Hessen B,Teuben J H.Switching a catalyst system from ethene polymerization to ethene trimerization with a hemilabile ancillary ligand[J].Angew Chem Int Ed,2001,40(13):2516-2519.
[20] Edwin Otten,Aurora A Batinas,Auke Meetsma,et al.Versatile coordination of cyclopentadienyl-arene ligands and its role in titanium-catalyzed ethylene trimerization[J].J Am Chem Soc,2009,131(14):5298-5312.
[21] Deckers P J W,Hessen B.Catalyst system for the trimerisation of olefins:WO,02/066404[P].2002-08-29.
[22] Deckers P J W,Hessen B.Catalyst system for the trimerisation of olefins:WO,02/066405[P].2002-08-29.
[23] Huang Jiling,Wu Tianzhi,Qian Yanlong.Ethylene trimerization with a half-sandwich titanium complex bearing a pendant thienyl group[J].Chem Commun,2003,39(22):2816-2817.
[24] Wu Tianzhi,Qian Yanlong,Huang Jiling.Catalytic trimerization of ethylene by half-sandwich titanium complexes bearing a pendant ethereal group[J].Journal of Molecular Catalysis A:Chemical,2004,214(2):227-229.
[25] Arno N J Blok,Peter H M Budzelaar,Anton W Gal.Mechanism of ethene trimerization at an ansa-(arene)(cyclopentadienyl)titanium fragment[J].Organometallics,2003,22(13):2564-2570.
[26] Yasuhiko Suzuki,Shinsuke Kinoshita,Atsushi Shibahara,et al.Trimerization of ethylene to1-hexene with titanium complexes bearing phenoxy-imine ligands with pendant donors combined with MAO[J].Organometallics,2010,29(11):2394-2396.
[27] Sugimura K,Nitabara T S,Fujita T.Olefin polymerization catalyst and polymerization of olefin using the same:JP,10324710[P].1998-12-08.
[28] Ban K,Hayashi T,Suzuki Y.Preparation of alpha-olefin multimer:JP,11060627[P].1999-03-02.
[29]BanK,HayashiT,SuzukiY.Productionofalpha-olefin oligomer:JP,10101587[P].1998-04-21.
(編輯:吳雅榮)
Research progress in non-chromium catalysts for ethylene trimerization
Sui Junlong
(Yanshan Branch of Beijing Research Institute of Chemical Industry,SINOPEC,Beijing102500,China)
This paper reviewedthe studies onfive types of non-chromium catalysts for ethylene trimerisation,including zirconium,vanadium,tantalum,titanium and nickel based catalysts in the light of the variety of the metallic elements in active center of the catalysts. The structural character of these catalysts, catalytic mechanism and reaction feature of ethylene trimerization in the presence of the catalysts were introduced.The effects of ligand structure,reaction temperature,pressure,cocatalyst amount on activity and selectivity towards1-hexene of the catalysts were compared.The titanium-based catalyst exhibits outstanding behavior among the catalysts for its excellent selectivity towards1-hexene as well as high activity;however,a very large consumption of methylaluminoxane as its cocatalyst is considered as a shortcoming of the catalytic system.
ethylene;trimerization;non-chromium catalyst;reaction mechanism
TQ426.92
A
1002-1396(2012)05-0070-05
2012-03-27。
修回日期:2012-06-26。
隋軍龍,1970年生,高級工程師,碩士,1993年畢業(yè)于天津大學化工系工業(yè)催化專業(yè),現(xiàn)從事有機化工研究工作。 E-mail:suijl.bjhy@sinopec.com;聯(lián)系電話:(010)69330674。
猜你喜歡
新世紀智能(數(shù)學備考)(2020年11期)2021-01-04 00:38:16
中國外匯(2019年17期)2019-11-16 09:31:14
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
現(xiàn)代企業(yè)(2015年1期)2015-02-28 18:43:18
新高考·高一物理(2014年1期)2014-09-18 01:26:07
應用化工(2014年3期)2014-08-16 13:23:50
