鄭氏比蜢線粒體基因組全序列的測定與分析
2011-12-25 08:04:06楊輝,黃原
Zoological Research 2011年4期
關鍵詞:結構
楊 輝, 黃 原
(陜西師范大學 生命科學學院, 陜西 西安 710062)
鄭氏比蜢線粒體基因組全序列的測定與分析
楊 輝, 黃 原?
(陜西師范大學 生命科學學院, 陜西 西安 710062)
采用長距PCR 擴增及保守引物步移法測定并注釋了鄭氏比蜢(Pielomastax zhengi)的線粒體基因組全序列。鄭氏比蜢的線粒體基因組全長15 602 bp, A+T 含量為71.8%, 37個基因位置與飛蝗的一致, 基因間隔序列共計10處47 bp, 間隔長度從1~20 bp不等; 有14對基因間存在52 bp重疊, 重疊堿基數在1~8 bp之間。蛋白質基因的起始密碼子均為昆蟲典型的起始密碼子ATN。ND5 基因使用了不完全終止密碼子T, 其余基因均為典型的TAA或TAG。除 tRNASer(AGN)的 DHU 臂缺失外, 其余21個tRNA 基因的二級結構均屬典型的三葉草結構, 但在鄭氏比蜢中有5個tRNA(tRNACys、tRNALys、 tRNAPhe、 tRNAProtRNAArg)基因變異較大, 無法采用常規算法預測出來, 表現在這5個tRNA 二級結構的TψC臂僅有3~4對配對堿基, tRNALys和tRNAArg的反密碼臂僅有4對配對堿基。預測的 lrRNA 二級結構總共有 6 個結構域(結構域Ⅲ缺失), 44 個莖環結構。預測的 srRNA 的二級結構包含3 個結構域, 30 個莖環結構。比較鄭氏比蜢、西藏飛蝗(Locusta migratoria tibetensis)和疑鉤額螽(Ruspolia dubia)rRNA二級結構后,發現鄭氏比蜢與西藏飛蝗的更相似。A+T 豐富區中存在一個被認為與復制及轉錄起始有關的 Ploy(T)結構。
蜢總科; 鄭氏比蜢; 線粒體基因組; RNA二級結構
后生動物線粒體基因組通常是編碼 37個基因(13個蛋白質基因、22個tRNA基因及2個rRNA基因)和1個控制區(也稱A+T豐富區)的細胞器基因組(Wolstenholme, 1992; Ding et al, 2007)。線粒體DNA具有多拷貝數、母系遺傳、突變率高、極少發生重組等特性, 已被廣泛地運用于分子進化、系統地理學和系統發育關系等方面的研究(William et al,2001)。根據 NCBI(www.ncbi.nlm.nih.gov/genomes/ORGANELLES/mztax_short.html)的統計, 截至2010年 11月, 已測出全線粒體基因組的昆蟲綱種類有223種, 其中直翅目昆蟲34種。由于測序的類群相對較少, 所選物種的特殊進化模式及系統樹中可能的長枝吸引等問題, 利用線粒體基因組數據推斷直翅目系統發育和進化地位還存在一定的困難(Gao et al, 2009)。另外, 已測的34種直翅目昆蟲大部分是螽斯總科和蝗總科的, 還缺乏蜢總科的代表種類, 因而增加這些類群中有關線粒體基因組全序列數據將有利于建立比較穩健的系統發育關系。
鄭氏比蜢(Pielomastax zhengi)屬于直翅目蜢總科(Eumastacoidea)枕蜢科(Episactidae)。蜢總科是一類較罕見而又原始的小型昆蟲, 其中一些種類還被認為是第三紀的孑遺種類。它們的大多數種類生活在熱帶和亞熱帶的灌木叢或林區內(Huang et al,2009)。本文測定并注釋了鄭氏比蜢線粒體基因組全序列, 并對其編碼的22個tRNA和大小亞基rRNA的二級結構做了預測和分析, 以期為直翅目昆蟲線粒體譜系基因組學提供新的數據資料。
1 材料和方法
1.1 標本采集及總DNA的提取
鄭氏比蜢標本于2009年8月采集于河南內鄉縣寶天曼自然保護區, 保存于無水乙醇。總DNA采用酚-氯仿抽提法:將后足股節肌肉置于 600 μL 勻漿緩沖液(0.01 mol/L Tris, pH 7.8; 5 mmol/L EDTA,5% SDS, 50 ng proteinase K/μL)中研磨, 65 ℃水浴消化 3~5 h。用平衡酚(pH 7.6~7.8)提純2次, 再用 CI(氯仿∶異戊醇=24∶1)提純一次。最后用?20 ℃預冷的 100%乙醇沉淀和 70%乙醇洗滌總DNA, 保存于?20 ℃。
1.2 PCR擴增及測序
本研究所采取的測序策略是長 PCR產物步移法測序, 如有測序效果不佳或不能夠測通的區域,再做 sub-PCR來補齊缺口。首先, 通過四對長距PCR(L-PCR)引物將線粒體基因組擴增為 4條相互重疊的片段(片段A、A2、B1和B2), 4個片段的上下游引物序列見表1(Liu et al, 2006), 長距PCR反應體系的總體積是 15 μL, 包括 10×反應緩沖液 1.5 μL、25 mmol/L MgCl22 μL、2.5mmol/L dNTPs 3 μL,10 μmol/L 上、下游引物各 2μL、5 U/μL LA-TaqDNA聚合酶0.15~0.35 μL、總DNA為模板1 μL。然后,加ddH2O至終體積15 μL。PCR反應循環條件如下:93 ℃預變性 2 min→(92 ℃10 s, 48 ℃ 30 s, 68 ℃8 min)共 15 個循環→(92 ℃ 10 s, 48 ℃30 s, 68 ℃8 min +20 s)共 15個循環→68 ℃ 7 min→4 ℃保存。其次, 用 12對保守引物以片段 A、A1、B1和 B2為模板再擴增320~3 175 bp的短片段, 具體信息見表2 (Simon et al, 2006)。短片段擴增體系總體積為25 μL, 包括 10×反應緩沖液 2.5 μL、25 mmol/L MgCl23 μL、2.5 mmol/L dNTPs 2 μL, 10 μmol/L 上、下游引物各4 μL、5 U/μL TaqDNA聚合酶0.15~0.35 μL和長片段產物(作為DNA模板)1 μL。然后,加ddH2O至終體積25 μL。PCR 反應程序為: 94 ℃2 min→(94℃×30 s+38~55 ℃×30 s+72 ℃×1 min)共 30 個循環→72 ℃×7 min→4 ℃ forever。最后, 通過12條sub-PCR產物測序將缺口補齊, 使基因組連接成環。

表1 長PCR擴增引物序列Tab. 1 Primer sequences for long PCR amplication
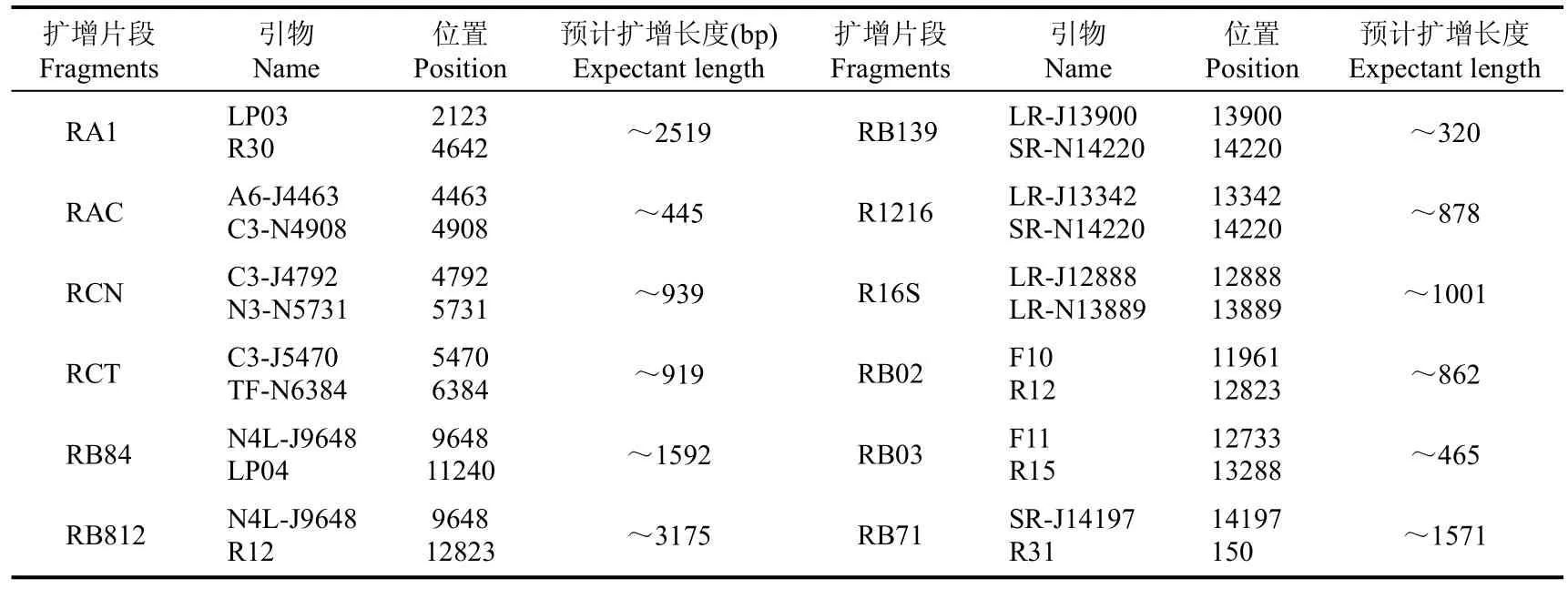
表2 補充擴增片段的引物對Tab. 2 The partial pair primers for the mitogenome complement fragment amplification
1.3 序列拼接、注釋和分析
使用Staden Package 1.7 (Bonfield et al, 1995)進行序列拼接和注釋, 通過與威廉劍角蝗Acrida_willemsei(NC_011303)線粒體 DNA序列進行比對確定蛋白質基因、tRNA基因和rRNA基因的位置,使用 MEGA 4.0 統計線粒體基因組堿基組成、蛋白質基因密碼子使用頻率等。使用 tRNAScan-SE ver.1.21進行 tRNA 二級結構預測。參考已發表的李氏大足蝗 srRNA二級結構和 lrRNA二級結構,對鄭氏比蜢線粒體基因組 srRNA和lrRNA的二級結構進行預測(Gao et al, 2009)。
2 結 果
2.1 線粒體基因組組成與基因排列
鄭氏比蜢線粒體基因組序列已提交到GenBank,登錄號為JF411955。本研究共測得41條可用序列,拼接后的序列總長度為 1 5618 bp, 序列覆蓋度1.98倍, 除去兩端重疊序列得到鄭氏比蜢線粒體基因組的總長度為 1 5602 bp。它由13個蛋白質編碼基因(protein-coding genes, PCGs)、22 個 tRNA 基因、2 個 rRNA 基因(lrRNA 和 srRNA)組成, 基因排列順序(圖 1)與蝗總科昆蟲的排列順序相同, 也存在tRNAAsp(D)和tRNALys(K)的倒置現象(Huang et al, 2010)。 基因排列非常緊密, 存在 14 處共 52 bp的基因重疊, 重疊區長度 1~8 bp 不等; 基因間隔序列總長度為47 bp, 共計10 處, 大小 1~20 bp之間, 基因間隔最大的在 tRNALys與 ATP8之間和ND5與 tRNAHis之間, 分別是 20 bp和10 bp; 既沒有重疊, 也沒有間隔的基因對共計 4 處。其中tRNAGln、tRNACys、tRNATyr、tRNAPhe、tRNAHis、tRNAPro、tRNALeu、tRNAVal和 ND1、ND4、ND4L、ND5 以及16s rRNA、12srRNA基因位于N鏈上, 其余23 個基因位于J 鏈上, 具體信息見表3。
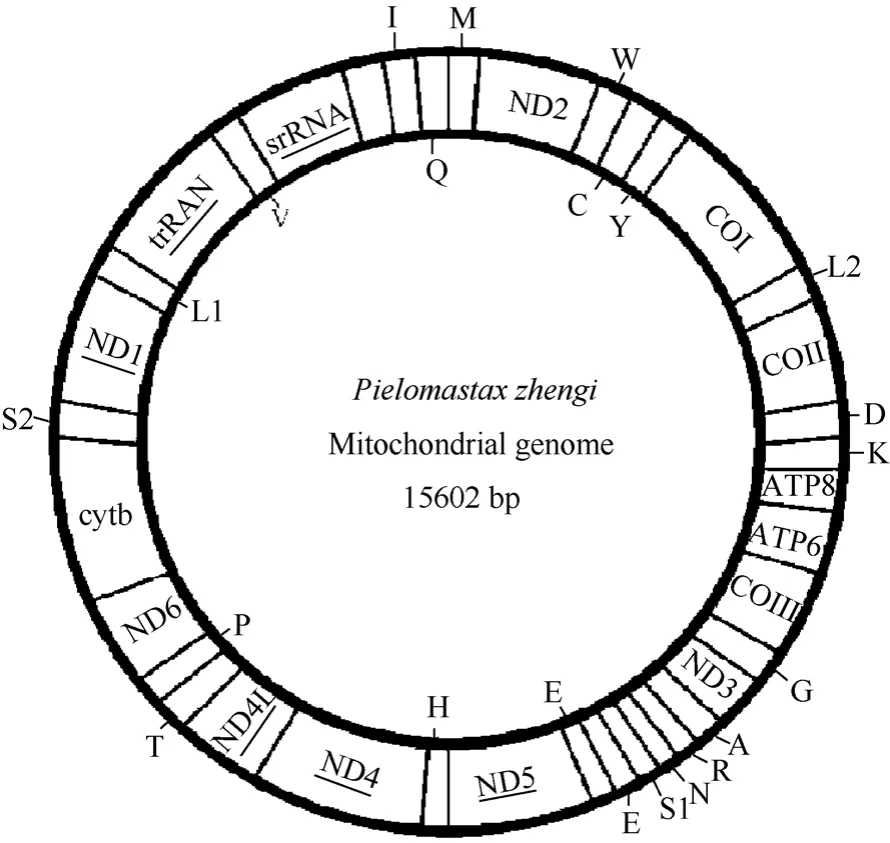
圖1 鄭氏比蜢線粒體基因組結構Fig. 1 The map of the Pielomastax zhengi mitogenome
2.2 線粒體基因組的堿基組成及密碼子使用
鄭氏比蜢線粒體基因組的堿基組成具有很強的AT 偏向性, 在4種堿基中, A含量最多, G含量最少。J鏈的堿基含量為: A(39.4%) > T(32.4%) >C(15.6) > G(12.6%), AT(71.8%)遠大于 CG(28.2%),這種現象普遍存在于蛋白質基因、tRNA和 rRNA基因, 尤其在A+ T 富集區。鄭氏比蜢的13個蛋白質基因最常用的起始密碼子是 ATG, 其次是 ATC和 ATT。除COⅢ、ND3、CytB和ND1以TAG作為終止密碼子, ND5的終止密碼子為不完全的T外,其 8個蛋白質基因的終止密碼子都為完整的 TAA,具體信息見表 3。鄭氏比蜢線粒體基因組兩條鏈上編碼的蛋白質密碼子使用情況存在差異, 即 J 鏈編碼基因偏向于使用第3位點為A的密碼子, 而N鏈偏向于使用第3位點為U的密碼子。
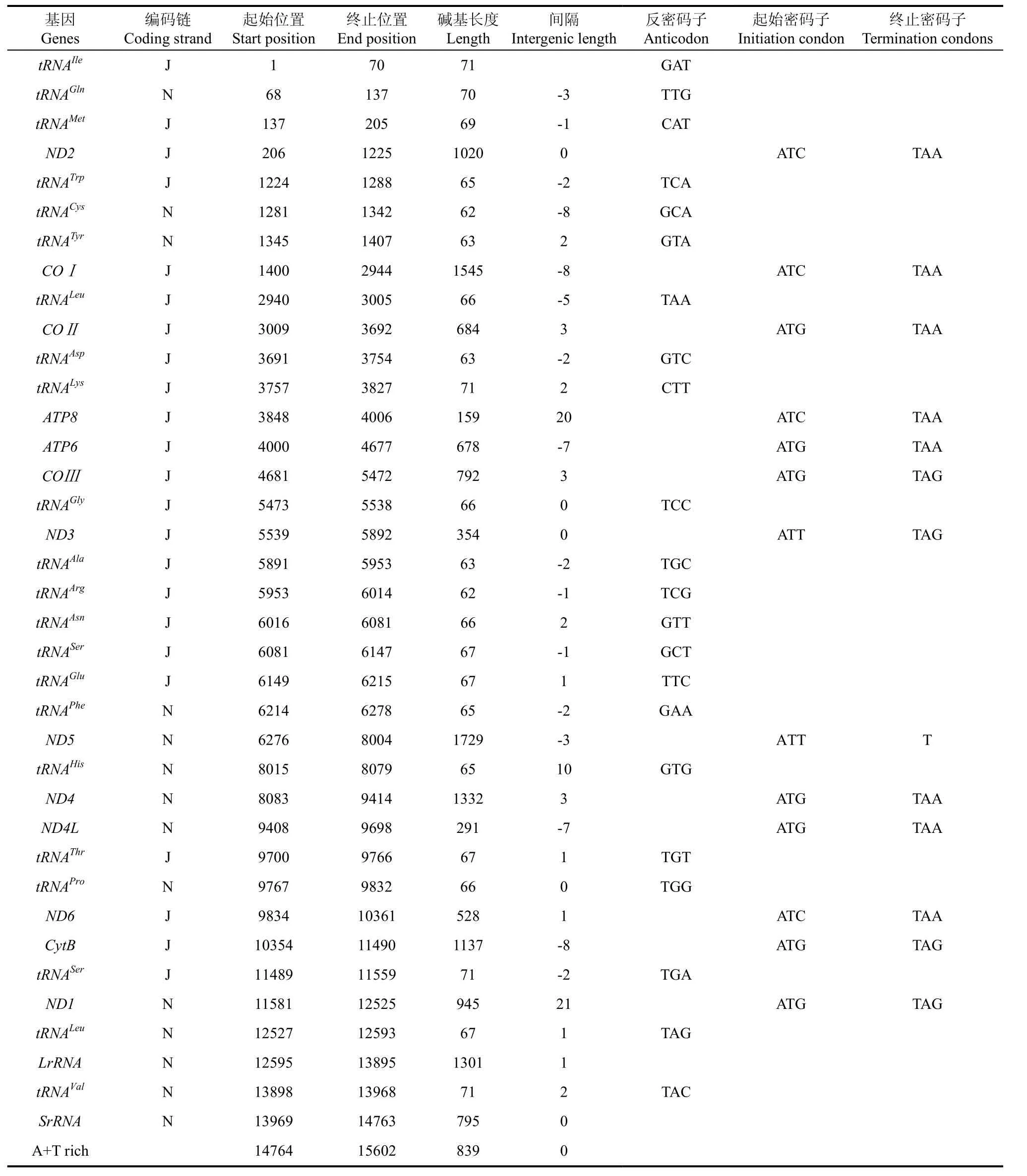
表3 鄭氏比蜢線粒體基因組組成Tab. 3 Gene organization of the Pielomastax zhengi mitochondrial genome
2.3 tRNA二級結構
鄭氏比蜢線粒體基因組有 22種 tRNA, 其中14種位于重鏈上, 8種位于輕鏈上。用 tRNAScan-SE 1.21 預測出鄭氏比蜢16個 tRNA 的二級結構,其余 6 個 tRNA 二級結構是通過與威廉劍角蝗Acrida willemsei(NC_011303)比對確定其在整個基因組上的位置后繪出。在所預測的22 種tRNA二級結構中總共出現了37處錯配, 其中有32對為GU錯配; 其余 5對錯配中:2對 UU 錯配發生在tRNAAla和 tRNAPhe的氨基酸接受臂和反密碼子臂;1對AC錯配發生在tRNAMet的氨基酸接受臂; 1對AA錯配發生在tRNALys的氨基酸接受臂; 1對 UC錯配發生在 tRNAPro的氨基酸接受臂。除了tRNAAsp、tRNAVal和tRNASerAGN(S)的反密碼環是9個堿基外, 其余的反密碼環均為7個堿基(圖2)。
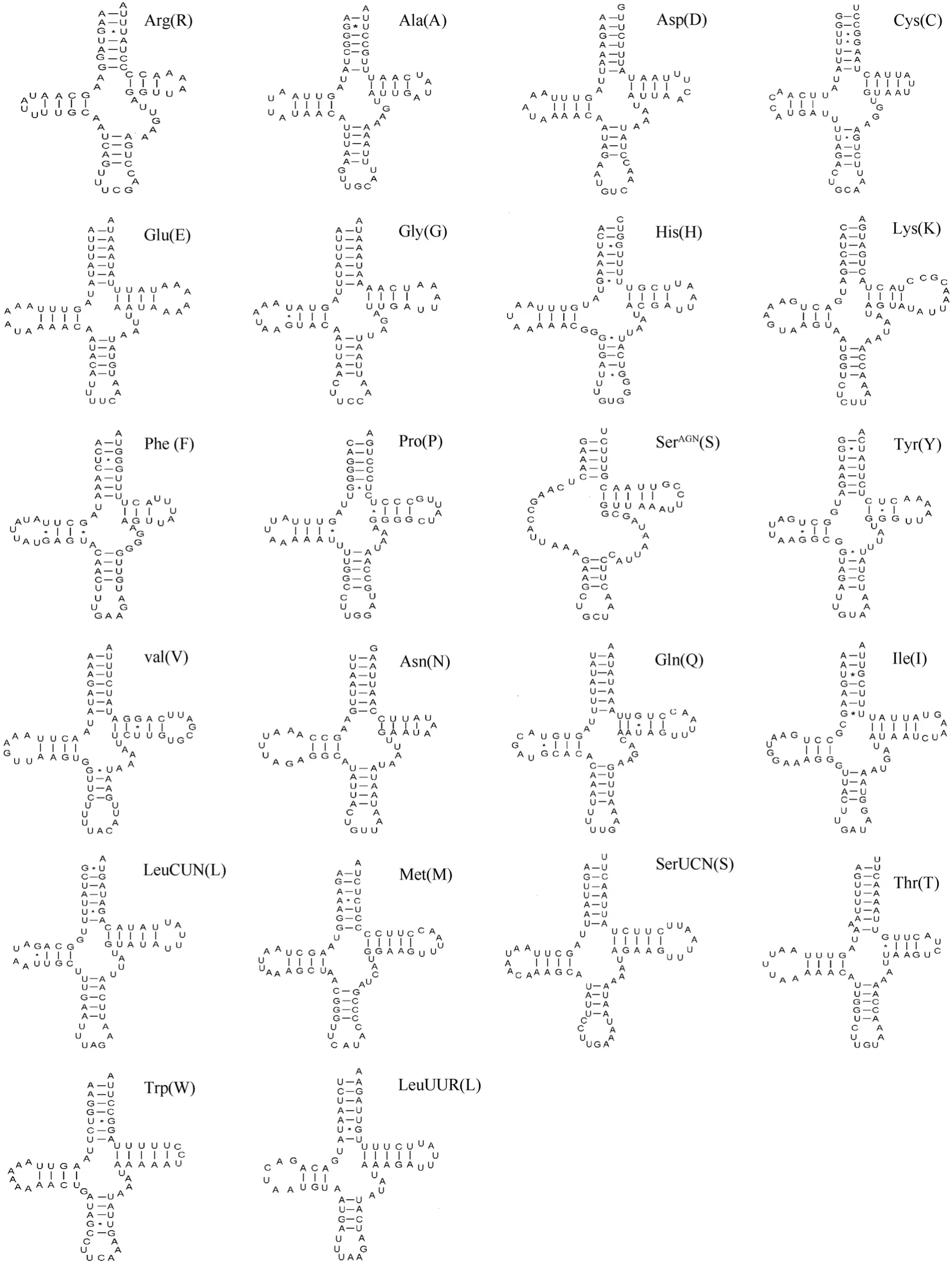
圖2 鄭氏比蜢線粒體基因組tRNA二級結構Fig. 2 The tRNA secondary structures of the Pielomastax zhengi mitogenome
2.4 rRNA二級結構
鄭氏比蜢線粒體基因組的lrRNA和srRNA分別位于tRNALeu(L-CUN)和tRNAVal(V), 以及tRNAVal(V)和D-loop之間, 其長度分別是1 301 bp和795 bp。兩個rRNA 基因的A+T含量為75.2%, 高于整個基因組的平均A+T含量(71.8%), G含量(15.1%)是C含量(9.8%)的近兩倍。以短額負蝗和西藏飛蝗的lrRNA和srRNA二級結構為基礎對鄭氏比蜢的二級結構進行了預測。結果顯示, 鄭氏比蜢 lrRNA二級結構總共有6個結構域(結構域Ⅲ缺失)和44個莖環結構。srRNA的二級結構包含3個結構域和30個莖環結構(圖2)。鄭氏比蜢的lrRNA和srRNA二級結構與短額負蝗和西藏飛蝗基本相似, 只在少數位置存在差異。
2.5 A+T豐富區
鄭氏比蜢的 A+T豐富區位于 srRNA與tRNAIle(I)之間, 全長 839 bp, A+T 含量為 82.1%,明顯高于全基因組的平均水平(71.8%)。在鄭氏比蜢的A+T豐富區發現一段 Poly(T)(T stretch), 且能形成一個莖環結構(圖4)。
3 討 論
3.1 基因排列順序


圖3 鄭氏比蜢rRNA(lrRNA和 srRNA)二級結構預測結果Fig. 3 The secondary structures of mitochondrial rRNAs (lrRNA and srRNA)of Pielomastax zhengi
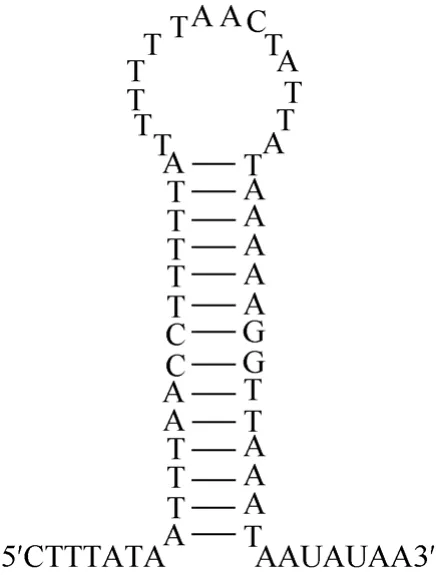
圖4 A+T富集區Poly(T)及附近序列所形成的莖環結構Fig. 4 Stem-loop structure at the poly- T stretch in the A+T rich region of Pielomastax zhengi mitogenome
在直翅目昆蟲線粒體基因組的研究中發現蝗亞目的蝗總科、蚤螻總科和蚱總科基因排列次序相同, 都發生了 KD 倒置現象, 即 tRNAAsp(D)排在tRNALys(K)基因上游(DK 排列) (Ballard & Dean,2001)。蜢總科的變色烏蜢為 KD 排列, 與蝗亞目其他總科不同, 而與多數螽亞目昆蟲的排序方式相同(Huang et al, 2010)。然而, 本研究所測得鄭氏比蜢的基因排列順序仍然與蝗總科的排列順序相同,即也發生了KD 倒置現象, 形成這種排列方式的原因是由于DK這二個基因所對應的 DNA片段發生了倒置。目前關于 DNA片段倒置產生的原理, 最有說服力的是Mark和Nick的觀點, 即雙鏈斷裂和重新結合模型(Zhong et al, 2005; Ye et al, 2008)。關于蜢總科昆蟲的線粒體基因組排列順序有三種可能:(1)大部分蜢總科線粒體基因組排列順序與蝗亞目的相同; (2)大部分蜢總科線粒體基因組排列順序與螽亞目的相同; (3)蜢總科昆蟲的線粒體基因組排列順序是隨機的。蜢總科昆蟲線粒體基因組排列究竟屬于那種可能, 還有待研究更多蜢總科物種的線粒體基因組進一步證實, 但偏向第一種情況的可能性更大, 因為蜢總科昆蟲無論從形態上分類還是從分子系統上劃分都表明與蝗亞目昆蟲的親緣關系更近。
3.2 tRNA二級結構
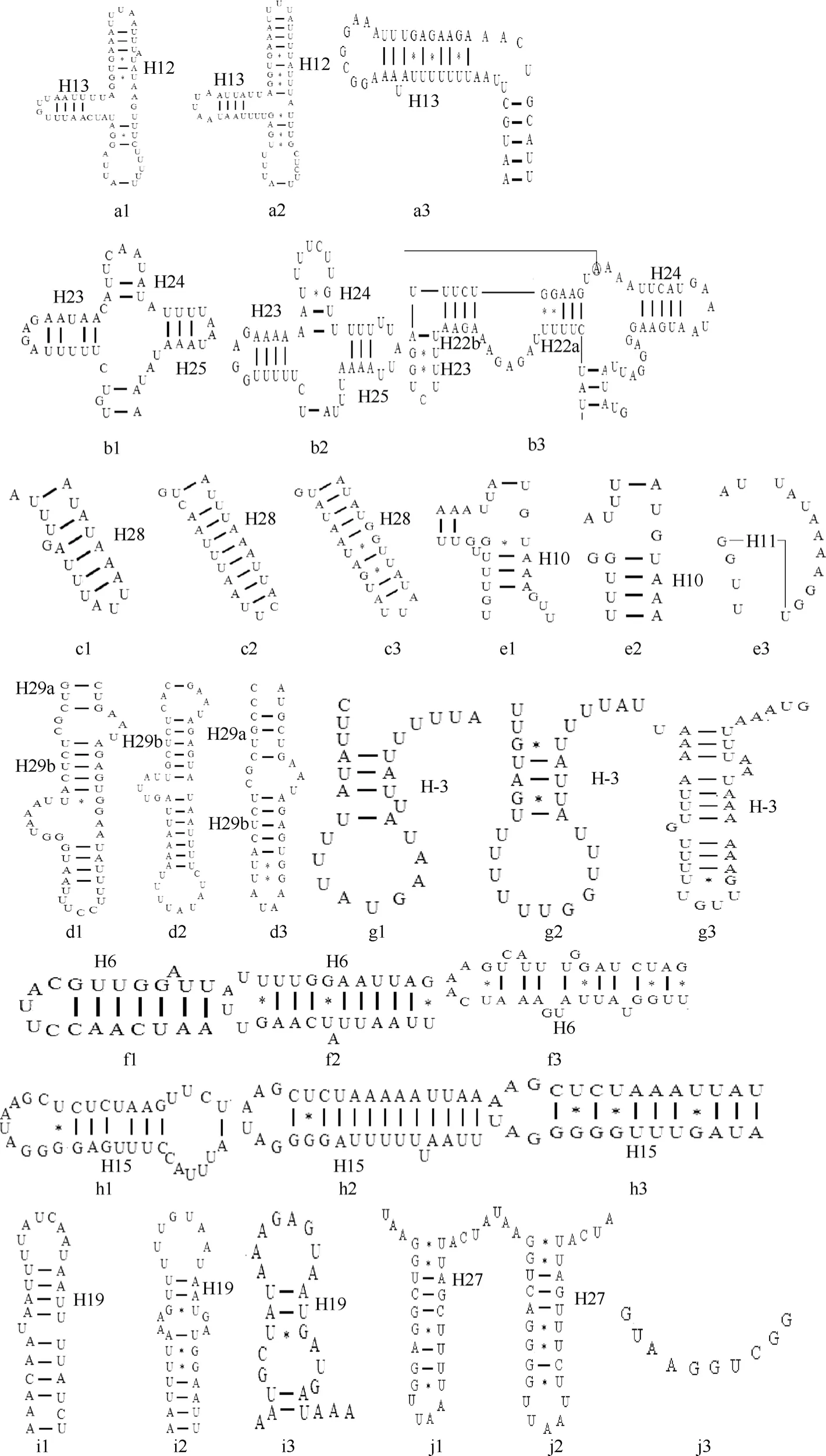
圖5 鄭氏比蜢、西藏飛蝗和疑鉤額螽rRNA二級結構的比較Fig. 5 The comparison of rRNA secondary structures among Pielomastax zhengi, Locusta migratoria tibetensis and Ruspolia dubia
大部分直翅目昆蟲線粒體基因組通過 tRNAScan-SE 1.21可以預測出20個tRNA基因。本研究的鄭氏比蜢只能預測出16個tRNA基因, 除了在大部分直翅目昆蟲都不能預測出來的 tRNAser(UCN)以外,還有5個tRNA 基因沒有預測出來。通過比較分析發現, 除了 tRNAser(UCN)不具有典型的三葉草二級結構外, 鄭氏比蜢其余的5個tRNA二級結構雖然具有典型的二級結構, 但是也存在一些差異, 即典型的三葉草結構的T¢C臂有5對配對堿基, 而這5個tRNA 二級結構的T¢C臂只有3~4對配對堿基; 另外, tRNALys和tRNAArg的反密碼臂只有4對配對堿基。這些結構特征可能是導致5個tRNA基因沒能預測出來的原因。另外, 預測的 22種tRNA二級結構中總共出現了 37處錯配, 其中 32對為GU錯配。有人認為線粒體基因組tRNA基因的部分錯配可以通過 RNA編輯校正, 不會引起氨基酸轉運上的障礙(Yokobori & P??bo, 1995; Dang et al, 2008)。
3.3 rRNA二級結構
rRNA二級結構模型對于理解核苷酸的替換模式和評估 rRNA系統發育信息的可靠性非常重要(Shi et al, 2008)。預測的鄭氏比蜢 lrRNA二級結構總共有 6 個結構域(結構域Ⅲ缺失)和44 個莖環結構(圖2 B); srRNA的二級結構包含 3 個結構域和30 個莖環結構(圖2 A)。lrRNA二級結構的結構域IV和V比較保守, srRNA二級結構的結構域I和II變異比較大。本文將鄭氏比蜢的rRNA二級結構與蝗亞目西藏飛蝗的rRNA二級結構和螽亞目疑鉤額螽的rRNA二級結構(Zhou et al, 2007; Jiang, 2010)進行了比較, 結果發現它們lrRNA的二級結構有四處存在明顯差異(圖3), srRNA的二級結構有6處存在明顯差異。由此可見lrRNA要比srRNA更保守一些。另外, 通過它們rRNA二級結構的比較發現,鄭氏比蜢rRNA二級結構與疑鉤額螽rRNA二級結構之間的差異明顯大于鄭氏比蜢rRNA二級結構與西藏飛蝗rRNA二級結構之間的差異(圖5)。因而,鄭氏比蜢與西藏飛蝗的親緣關系可能更近, 也就是與蝗亞目昆蟲的親緣關系更近。
3.4 A+T富集區
目前對 A+T 富集區域的研究關注主要集中于所包含的復制相關的調控信息。現在公認的是昆蟲的線粒體 N鏈的復制起點(ON)位于控制區內, J鏈的復制則是在輕鏈的復制完成97%之后才開始, 即認為OJ(J 鏈的復制起點)位于距離ON97% mtDNA長度的位置上(Goddard & Wolstenholme, 1978; Saito et al, 2005)。Saito et al(2005)以果蠅為主要研究對象,同時也對10個目的20種昆蟲的A+T富集區進行了研究。根據實驗結果, 前期的相關研究認為, 由于ON位于控制區內, 加之其上游的一段 Poly(T)結構,它可能與復制起點的識別有關(ye et al, 2008)。本研究所分析的鄭氏比蜢 A+T 富集區也有那樣一段Poly(T)結構, 應該也與復制起點的識別有關。
Ballard JWO, Dean MD. 2001. The mitochondrial genome: mutation,selection and recombination[J].Curr Opin Genet Dev, 11: 667-672.
Bonfield JK, Smith KF, Staden R. 1995. A new DNA sequence assembly program [J].Nucleic Acids Res, 24: 4992-4999.
Dang JP, Liu N, Ye W, Huang Y. 2008. Complete mitochondrial genome sequence ofGastrimargus marmoratus(Thunberg) (Orthoptera :Acridoidea) [J].Acta Entomol Sin, 51(7): 671-680. [黨江鵬, 劉 念,葉 偉, 黃 原. 2008. 云斑車蝗線粒體基因組全序列測定與分析.昆蟲學報, 51(7): 671-680.]
Ding FM, Shi HW, Huang Y. 2007. Complete mitochondrial genome and secondary structures of lrRNA and srRNA ofAtractomorpha sinensis(Orthoptera, Pyrgomorphidae) [J].Zool Res, 28(6): 580-588. [丁方美,師紅雯, 黃 原. 2007. 短額負蝗線粒體基因組及其 lrRNA 和srRNA 二級結構分析. 動物學研究, 28(6): 580-588]
Gao J, Cheng CH, Huang Y. 2009. Analysis of complete mitochondrial genome sequence ofAeropus licentiChang [J].Zool Res, 30(6):603-612. [高佳, 程春花, 黃原. 2009. 李氏大足蝗線粒體全基因組序列分析. 動物學研究, 30(6):603-612].
Goddard JM, Wolstenholme DR. 1978. Origin and direction of replication in mitochondrial DNA molecules fromDrosophila melanogaster[J].Proc Natl Acad Sci USA, 75(8): 3886-3890.
Huang Y, Liu N, Lu HM, 2010. Research progress in mitochondrial genomes of the Orthoptera insects [J].Acta Entomol Sin, 53(5) :581-586. [黃原, 劉念, 盧慧甍. 直翅目昆蟲線粒體基因組研究進展.昆蟲學報, 53(5) : 581-586.]
Huang JH, Huang Y, Zhou SY. 2009. Check list of Chinese species of superfamily Eumastacoidea (Orthoptera: Caelifera) [J].J Guangxi Normal Univ, 27(1): 84-87. [黃建華, 黃 原, 周善義. 2009. 中國蜢總科昆蟲名錄. 廣西師范大學學報, 27(1): 84-87.]
Jiang DY. 2010. Determination and analysis of four complete mitochondrial genome sequences of locust populations [D]. College of Life Sciences,Xi’an Shaanxi Normal University. [江東洋. 2010. 中國四種飛蝗線粒體基因組序列的測定與分析 [D]. 西安: 陜西師范大學生命科學學院.]
Liu N, Hu J, Huang Y. 2006. Amplification of grasshoppers complete mitochondrial genomes using long PCR [J].Chn J Zool, 41: 61-65.[劉念, 胡 靖, 黃 原. 2006. 應用長 PCR 擴增蝗蟲線粒體全基因組.動物學雜志, 41: 61-65.]
Saito S, Tamura K, Aotsuka T. 2005. Replication origin of mitochondrial DNA in insects[J].Genetics, 171(4): 1695-1705.
Shi HW, Ding FM, Huang Y. 2008. Complete sequencing and analysis of mtDNA inPhlaeoba albonemaZheng [J].Chn J Biochem Mol Biol, 24(7): 604-611.
Simon C, Buckley TR, Frati F, Stewart JB, Beckenbach AT. 2006.Incorporating molecular evolution into phylogenetic analysis, and a new compilation of conserved polymerase chain reaction primers for animal mitochondrial DNA [J].Annu Rev Ecol Evol Syst, 37:545–579.Wolstenholme DR, 1992. Animal mitochondrial DNA: structure and evolution [J].Int Rev Cytol, 141: 173-216.
Yokobori S, P??bo S. 1995. Transfer RNA editing in land snail mitochondria [J].Proc Natl Acad. Sci USA, 92: 10432-10435.
Ye W, Dang JP, Xie LD, Huang Y. 2008. Complete mitochondrial genome ofTeleogryllus emma(Orthoptera: Gryllidae) with a new gene order in Orthoptera [J].Zool Res, 29(3): 236-244. [葉 偉, 黨江鵬, 謝令德,黃 原. 2008. 黃臉油葫蘆線粒體基因組:一種新的基因排列方式.動 物 學 研 究, 29(3):236-244.]
Zhong J, Li G, Liu ZQ, Li QW, Wang YQ. 2005. Gene rearrangement of mitochondrial genome in the vertebrate [J].Acta GenetSin, 32(3):322-330. [鐘 婧, 李 光, 劉忠權, 李慶偉, 王義權. 2005. 脊椎動物線粒體DNA 的基因重排. 遺傳學報, 32 (3): 322-330.]
Zhou ZJ, Huang Y, Shi FM. 2007. The mitochondrial genome ofRuspolia dubia(Orthoptera: Conocephalidae) contains a short A+T-rich region of 70 bp in length [J].Genome, 50: 855-866.
Analysis of the complete mitochondrial genome sequence ofPielomastax zhengi
YANG Hui, HUANG Yuan?
(School of Life Sciences,Shaanxi Normal University,Xi’an710062,China)
The complete mitochondrial genome sequence ofPielomastax zhengiwas determined by using long PCR and conserved primer walking approaches. The results showed that the entire mitochondrial genome ofPielomastax zhengiis 15 602 bp long with A+T content of 71.8%. All 37 genes are conserved in the positions observed in those ofLocusta migratoria. All the genes are closely assembled by leaving 10 intergenic spacers in between. Those intergenic spacers are 47 bp in total (excluding the A+T rich region), with individual size ranges from 1 bp to 20 bp. In addition,there are a total of 52 bp overlapping sequences among 14 genes, ranging from 1?8 bp. All protein-coding genes start with a typical initiation codon in insects, ATN. Twelve protein-coding genes use the usual TAA and TAG termination codons, whereas, the ND5 genes have an incomplete termination codon (T). Except the tRNASer (AGN), whose DHU arm is absent; all the other 21 tRNA genes have typical clover-leaf secondary structures. But inPielomastax zhengi, the secondary structures of five tRNA (tRNACys, tRNALys, tRNAPhe, tRNAPro, tRNAArg) genes can not be predicted by the conventional methods as they have only 3?4, rather than 5 base pairs in the TψC arm, while, the tRNALysand tRNAArghave only 4 base pairs in the anticode arm. The predicted secondary structures of lrRNA and srRNA have 6 domains with 44 helices and 3 domains with 30 helices, respectively. The results of the rRNA secondary structure comparison showed thatPielomastax zhengiis more closely related withLocusta migratoria tibetensisthanRuspolia dubia. Like most insects, the mitochondrial genome ofPielomastax zhengihas a non-coding A+T rich region containing a polythymidine stretch, which may be involved in the replication and/or translation initiation of other genes.
Eumastacoidea;Pielomastax zhengi; Mitochondrial genome; RNA secondary structure
Q969.26; Q754
A
0254-5853-(2011)04-0353-10
10.3724/SP.J.1141.2011.04353
2011-01-06;接受日期:2011-03-28
自然基金資助項目(30670279;30970346)
?通訊作者(Corresponding author),E-mail: yuanh@snnu.edu.cn
楊輝,碩士研究生,研究方向:基因組學
猜你喜歡
小獼猴智力畫刊(2023年4期)2023-04-23 08:49:58
哲學評論(2021年2期)2021-08-22 01:53:34
中華詩詞(2019年7期)2019-11-25 01:43:04
模具制造(2019年3期)2019-06-06 02:10:54
中學生數理化·高一版(2018年1期)2018-02-10 05:20:03
影視與戲劇評論(2016年0期)2016-11-23 05:26:01
七彩語文·寫字與書法(2016年7期)2016-07-28 21:40:22
七彩語文·寫字與書法(2016年6期)2016-07-15 19:36:34
人間(2015年21期)2015-03-11 15:23:21
現代企業(2015年9期)2015-02-28 18:56:50

- Zoological Research的其它文章
- Localization of stationary pronuclei during conjugation of Paramecium as indicated by immunofluorescence staining
- 一種有效區分移植細胞和宿主細胞腦損傷模型的建立
- 懸尾應激對小鼠空間記憶及其反轉學習的損傷效應
- Metabolism and thermoregulation between Mrs Hume’s Pheasant (Syrmaticus humiae) and Elliot’s Pheasant (S. ellioti)
- Notch signaling dependent differentiation of cholangiocyte-like cells from rhesus monkey embryonic stem cells
- Afferent and efferent pathways in the visual system of the freshwater snail Planorbarius corneus