哈密煤惰質組結構模型的分子模擬
2011-11-10 03:04:28趙凱榮樊海治
山西煤炭 2011年7期
趙凱榮,樊海治
(1太原理工大學 礦業工程學院,山西 太原030024;2內蒙古煤田地質局109隊,內蒙古 海拉爾 021008)
哈密煤惰質組結構模型的分子模擬
趙凱榮1,樊海治2
(1太原理工大學 礦業工程學院,山西 太原030024;2內蒙古煤田地質局109隊,內蒙古 海拉爾 021008)
采用計算機分子模擬技術,在分子尺度上研究了哈密惰質組結構性質。首先采用分子力學、退火動力學模擬方法得到優化后三維結構圖形;經過計算后對比分子勢能變化,發現范德華力是分子中的主要作用力。在量子化學計算基礎上,進一步對其鍵級、鍵長等結構參數進行了分析。
惰質組;分子模擬;微觀結構
煤分子結構的研究一直是煤科學領域的前沿課題。科研工作者們[1,2]從各種角度提出了不同的結構模型,它們反映了煤的一些特征以及當時研究的水平,對探索煤結構本質提供了有益信息。然而這種建立在經驗和假設基礎上的模型不能準確地反映煤的結構特性,在此基礎上進行煤結構和反應性的關聯性研究更是不可能[3]。隨著科技的發展,建立在分子力學、分子動力學基礎上的計算機輔助分子設計已廣泛應用于煤科學研究領域(煤結構和性質),使得從三維的角度建立和模擬煤大分子立體結構變得容易[4-6],也使得在分子水平上討論煤結構與性質、反應機理成為可能[7]。該技術可以觀察到原子運動過程中各種微觀細節,能為我們提供關于分子體系的微觀理解,是對理論計算和實驗的有力補充。
作為后備能源基地的西部吐哈盆地發育有巨厚的中下侏羅統煤系,本文以新疆哈密惰質組顯微組分為研究對象。Daniel Van Niekerk[8]基于模擬實驗構建了一個惰質組豐富的南非二疊紀煤炭模型。國外高惰質組煤(岡瓦納煤)主要分布于澳大利亞、印度等國的一些二疊紀地層中,但其在成煤時代、煤階、礦物分布等方面與我國西部侏羅紀煤有很大差異[9]。本文在實驗分析的基礎上,采用分子模擬手段深入研究哈密惰質組分性質,進一步科學認識西部弱還原煤。
1 計算方法和過程
首先基于13C CP/MAS NMR測試結果和HI的元素分析,計算出樣品的結構參數,在此基礎上構建了HI的初始化學結構模型。利用ACD/CNMR preditor軟件對HI的初始結構模型進行化學位移計算獲得了模擬13C NMR譜,根據模擬譜和實驗譜的對比結果,對HI的初始模型進行了修正,最終得到與實驗結果吻合的化學結構模型。
把在ACD/Chemsketch繪圖軟件里搭建起的哈密惰質組結構模型,見圖1,導入到服務器中的Materials studio 4.1模擬軟件,對結構模型進行加氫飽和,然后利用Clean工具對結構模型進行初步優化。
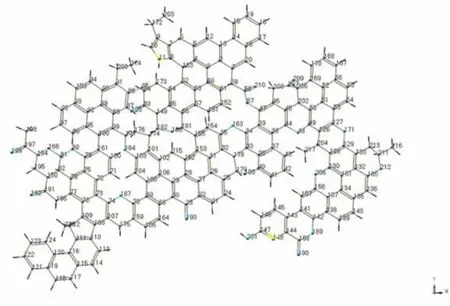
圖1 計算前HI單元體幾何構形
分子力學和分子動力學計算都使用Materials studio 4.1(MS)軟件包內的MS.Forcite計算模塊,所有的模擬過程中均采用Dreiding力場。分子力學參數設置為:最陡下降法(SteepesTMinimizer),迭代步數為5000,收斂標準采用Medium,其中原子均方根力(RMS Force)的標準為0.1kcal/mol·A,能量偏差
退火動力學模擬的初始溫度設置為300K,最高溫度設為600K,升溫速率為60K/次,每段都進行NVT的分子動力學模擬,模擬時間為20ps,控溫程序選擇Nose方法,退火模擬的循環次數設置為10。分子動力學模擬后最優幾何構型,見圖2。
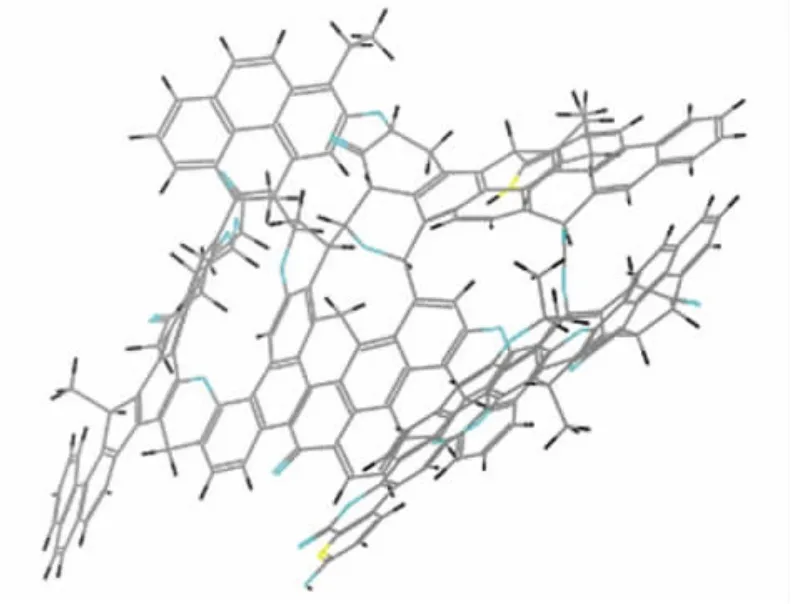
圖2 HI結構模型的能量最優幾何構型
將優化后獲得的分子最優構象圖2放入Materials studio 4.1中的Vamp模塊中進行計算。Vamp是半經驗量子化學的分子軌道軟件包,設置如下:計算方法選用NDDO中的AM1方法,收斂標準選擇Medium,在性質計算中選上頻率分析(Frequency),旋轉選擇UHF,其它參數默認。
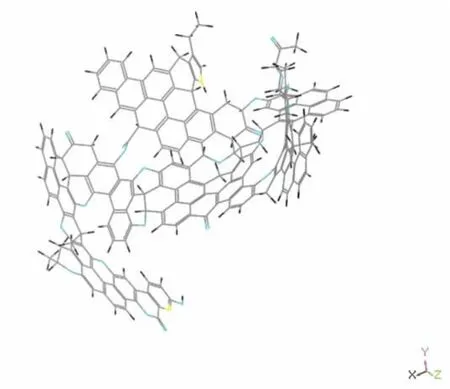
圖3 HI的半經驗計算后能量最小化構型
2 結果與分析
2.1 分子力學計算結果
經過分子力學和動力學計算后得到的HI的三維能量最小化構型,如圖2所示。相比較圖1,優化后的結構模型發生了明顯的彎曲變形,具有良好的立體感。HI的基本結構單元模型中含有三或四個苯環為主,優化后結構中芳香片層近乎平行。陳皓侃等[3]發現能量最小化煤分子構型中,芳香環傾向于以平行或重疊的形式排列,推測可能是環的吸引旋轉而形成的。團聚的分子模型已存在類石墨化現象,李軍等人[10]也發現:弱還原煤聚集態的結構呈片狀。
分子動力學模擬過程是分子構型隨著時間變化的動態過程,其中總勢能、成鍵相互作用能及非鍵相互作用能也呈一定的變化趨勢。在模擬過程中,模型分子快速團聚為更緊湊的結構,分子的總勢能隨時間增加而急速下降,隨后時間推移,最后階段模型總勢能基本保持恒定。
表1所列HI最終結構模型的勢能組成:非成鍵能EN比成鍵能(EV)高,是勢能的主要組成部分,說明結構模型的穩定性主要來自于非成鍵能的貢獻;非成鍵能中占主要地位的是范德華能,它的貢獻遠大于氫鍵力和靜電力,說明它是芳香片層結構的主要推動力。
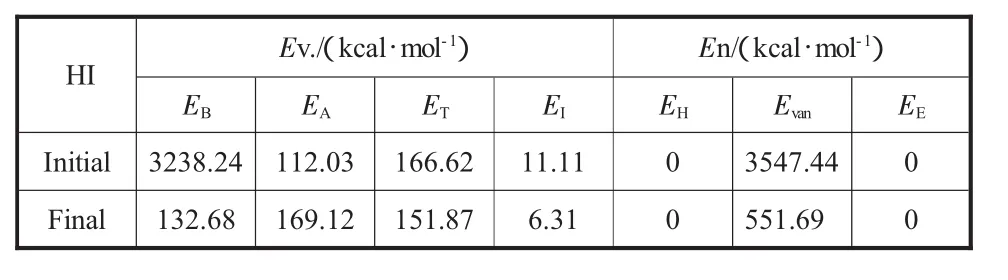
表1 HI結構模型經過能量最小化后的勢能變化
2.2 鍵級分析
通常鍵級越大,鍵的強度越大。表2中列出了對HI分子進行優化后所得到的鍵長、鍵級參數。經統計得出,HI結構模型中C194-C197、C83-C84、C58-C59、C207-C205、C83-C149處的鍵級比較弱,鍵級范圍在0.896-0.929間,并且都是和羰基碳原子相連的C-C單鍵。煤中氧的存在形式對煤的反應活性有很大的影響[5],這些鍵中由于受到羰基的影響,化學鍵的活性增強,在反應中更容易斷裂。Car—O和 Cal—O醚鍵的鍵能則相差較大:O199-C197、C191-O192、C207-O209、、C83-O125
鍵 級 最 高 , 范 圍 在 1.912-1.92;C211-O212、C179-O180、C177-O184鍵級比較弱,范圍在0.939-0.948。由此可見,同一結構中Car—O的鍵能較Cal—O的約高1數量單位,因此在受熱過程與脂肪碳相連的C—O類醚鍵更容易斷裂分解,也說明芳香結構位對反應活存在影響。
3 結論
本文采用分子模擬研究了HI結構和物理化學性質。首先采用分子力學、動力學的模擬方法,發現優化后三維HI結構呈片狀,具有較多的敞開式孔,芳香層片在空間規則的近平行排列,整體趨向于“石墨化”。分子力學、動力學計算結果表明,范德華力是分子中的主要作用力。
應用半經驗量子化學AM1方法對鍵長、鍵級等的計算結果表明:脂肪C—C鍵長較芳香C—C鍵長長,在受熱過程中更容易斷裂;結構的反應活性受羰基影響較大;邊緣C原子易于發生氧化反應,而芳香結構很穩定,很難反應。
[1]Given PH·The distribution oFhydrogen in coals and its relation to coal structure[J].Fuel,1960,39:147-153.
[2] Shinn,J H·From coal to single-stage and two-stage products:a reactive model oFcoal structure[J].Fuel,1984,63:1187-1196.
[3]陳皓侃,李保慶,李文.分子力學和分子動力學方法研究不同變質程度煙煤的分子結構[J].燃料化學學報.2000,28(5):459-462.
[4] Carlson G A,GranofFB·Modeling oFcoal structure using computer-aided molecular design[J].Am Chem Soc Div Fuel Chem Pre-prints,1989,34(3):780-786.
[5] Faulon J L,Carlson GA,Hatcher P G·Statistical models for bituminous coal:a three-dimensional evaluation oFstructural and physi-cal properties based on computer-generated structures[J]·Energy Fuels,1993,7(6):1062-1072.
[6]侯新娟,楊建麗,李永旺.煤大分子結構的量子化學研究[J].燃料化學學報,1999,27(增刊):142-148.
[7]孫慶雷,李文,陳皓侃,等.煤顯微組分分子結構模型的量子化學研究[J].燃料化學學報,2004,32(3):282-286.
[8] Daniel Van Niekerk,Jonathan P.Mathews.Molecular representations oFPermian-aged vitrinite-rich and inertinite-rich South African coals[J].Fuel,2010,89(1):73-82.
[9]白向飛,李文華,陳文敏,等.我國西部弱還原程度煤分布及煤質特征研究[J].煤炭學報,2005,30(4):502-506.
[10] 李軍,馮杰,李文英,等.強弱還原煤聚集態對其可溶性影響的分子力學和分子動力學分析[J].物理化學學報,2008,24(12):2297-2303.
Molecular Simulation oFHumul Coal Inertinite Structure Model
ZHAO Kai-rong1,FAN Hai-zhi2
(1.College oFMining Engineering,Taiyuan University oFTechnology,Taiyuan Shanxi 030024;2.No.109Team oFInner Mongolia Coalfield Geology Bureau,Hailar Inner Mongolia 021008)
The paper uses computer molecular simulation technology and studies the Humul coal inertinite structure on the molecular scale.First,the molecular mechanics and annealing kinetics simulation achieves the 3dimensional structure graphs.Molecular potential change is compared after the calculation;we find thaTVan Der Walls force is the main inter-molecular force.On the basis oFquantum chemical calculation,the bond order,length and other structure parameters are further analyzed.
inertinite;molecular simulation;microscopic structure
TQ530
A
1672-5050(2011)07-0035-03
2011-05-05
趙凱榮(1983—),男,山西聞喜人,在讀碩士研究生,主要從事煤田地質研究工作。(Energy Difference)為0.001kcal/mol,分子力場的電荷分布由電荷平衡法(QEq)獲得原子的凈電荷。
徐樹文
猜你喜歡
童話王國·奇妙邏輯推理(2024年5期)2024-06-19 16:03:38
哲學評論(2021年2期)2021-08-22 01:53:34
中學生數理化·七年級數學人教版(2020年10期)2020-11-26 08:24:50
數學物理學報(2020年2期)2020-06-02 11:29:24
中華詩詞(2019年7期)2019-11-25 01:43:04
模具制造(2019年3期)2019-06-06 02:10:54
影視與戲劇評論(2016年0期)2016-11-23 05:26:01
光學精密工程(2016年6期)2016-11-07 09:07:19
核科學與工程(2015年4期)2015-09-26 11:59:03
現代企業(2015年9期)2015-02-28 18:56:50
