TiO2/SiO2催化苯酚與碳酸二乙酯鄰位烷基化反應
2011-11-09 00:44:00呂列超李永昕
石油化工 2011年4期
關鍵詞:催化劑
呂列超,薛 冰,許 杰,李永昕
(常州大學 石油化工學院,江蘇 常州 213164)
TiO2/SiO2催化苯酚與碳酸二乙酯鄰位烷基化反應
呂列超,薛 冰,許 杰,李永昕
(常州大學 石油化工學院,江蘇 常州 213164)
采用等體積浸漬法制備了一系列TiO2/SiO2催化劑,通過XRD、NH3-TPD和Py-IR等手段對催化劑進行了表征;在連續流動固定床反應器上,考察了TiO2負載量、反應溫度、碳酸二乙酯(DEC)與苯酚的摩爾比及重時空速(WHSV)對苯酚與DEC鄰位烷基化反應的影響。實驗結果表明,L酸中心是苯酚鄰位烷基化反應的活性中心;隨TiO2負載量的增加,苯酚的轉化率逐漸增加,鄰乙基苯酚的選擇性先增加后減小,而2,6-二乙基苯酚的選擇性則逐漸增加,這主要是由負載TiO2后催化劑的酸中心數目增多引起的。以9%(w)TiO2/SiO2為催化劑,在360 ℃、0.42 MPa、n(DEC)∶n(苯酚)=1.0、WHSV=2 h-1、8 h 的適宜反應條件下,苯酚的轉化率為75.4%,鄰乙基苯酚和2,6-二乙基苯酚的選擇性分別為60.2%和16.3%。
二氧化鈦/二氧化硅催化劑;碳酸二乙酯;苯酚;鄰乙基苯酚;2,6-二乙基苯酚
苯酚的乙基化反應是一類重要的有機反應,其產物鄰乙基苯酚可用于制造酚醛樹脂、橡膠防老化劑和表面活性劑等;2,6-二乙基苯酚則是一種重要的醫藥中間體[1-2]。傳統的鄰乙基苯酚生產是從煤焦油中分離提取的,由于存在資源有限、工藝過程復雜等問題,該法已逐漸被化學合成法取代。化學合成法主要包括鄰乙基苯胺重氮化水解法、苯酚-乙烯烷基化法和苯酚-乙醇烷基化法。其中苯酚-乙醇烷基化法具有工藝簡單、環境友好等優點,是目前催化領域中的一個研究熱點[1,3],但乙醇作為烷基化試劑活性較低[4]。碳酸二乙酯(DEC)作為綠色化工原料,具有低毒、無腐蝕性和高化學活性等特點[5-6]。本課題組曾報道了苯與DEC合成乙苯的研究結果[7],DEC的活性遠遠高于乙醇。關于苯酚與DEC烷基化反應合成烷基苯酚的研究,迄今國內外鮮見報道。
傳統的苯酚烷基化催化劑(無水三氯化鋁、三氟化硼、液體酸和離子交換樹脂等)存在著腐蝕和污染嚴重、分離困難、易失活等缺點,使烷基苯酚的生產受到了制約[8]。由于固體酸催化劑具有高活性、催化劑可循環再生以及分離過程簡單等優點,在苯酚烷基化反應中,固體酸正在逐漸取代傳統的苯酚烷基化催化劑[9]。
本工作以DEC為乙基化試劑、TiO2/SiO2為催化劑,對苯酚與DEC鄰位烷基化反應進行了研究。
1 實驗部分
1.1 試劑
鈦酸丁酯:化學純,國藥集團化學試劑有限公司;苯酚、甲苯:分析純,國藥集團化學試劑有限公司;DEC:分析純,天津市化學試劑研究所;SiO2:工業級,青島海洋化工有限公司。
1.2 催化劑的制備
以SiO2為載體,采用等體積浸漬法在相同條件下制備一系列不同負載量的TiO2/SiO2催化劑。具體制備方法:將SiO2在120℃下干燥3 h,除去其中的水分備用;稱取一定量的鈦酸丁酯溶于適量的甲苯中,將所得溶液均勻浸漬于一定量的SiO2上,室溫下放置24 h,水浴蒸干甲苯,120℃下干燥3 h,以10℃/min的速率升溫至550℃焙燒4 h,即得TiO2/SiO2催化劑,記為wTiO2/SiO2,其中w表示TiO2的負載量(相對于催化劑的質量分數)。
1.3 催化劑的表征
采用理學公司D/Max 2500PC型X射線衍射儀對試樣進行 XRD表征,CuKα射線,掃描范圍2θ=5 ~80°,管電壓 40 kV,管電流 100 mA;采用Quantachrome公司CHEMBET-3000型化學吸附儀對試樣進行NH3-TPD表征;吡啶吸附紅外光譜(Py-IR)表征在Bruker公司TENSOR 27型傅里葉變換紅外光譜儀上進行,將試樣研細后壓制成自撐片,在380℃下抽真空處理2 h,冷卻至室溫后吸附吡啶,升溫脫附,記錄200℃時的Py-IR譜圖。
1.4 催化劑的性能評價
在氣相連續流動固定床反應器上對催化劑進行評價。將一定量的催化劑裝填于反應管中部,反應管兩端填充玻璃珠。采用雙柱塞微量泵進料,原料苯酚與DEC的混合液經預熱后由N2載入反應管,自上而下流經催化劑床層進行反應。反應后的產物用冷阱冷凝,定時收集液相產物進行氣相色譜分析。采用山東魯南瑞虹化工儀器有限公司SP-6890型氣相色譜儀進行分析,FFAP毛細管柱,FID檢測,采用歸一化法定量。
2 結果與討論
2.1 TiO2負載量對催化劑性能的影響
TiO2負載量對苯酚與DEC鄰位烷基化反應的影響見圖1。由圖1可見,苯酚的轉化率隨TiO2負載量的增加而增大,當TiO2負載量從3%增至12%時,苯酚的轉化率大幅上升;進一步增加TiO2負載量,苯酚的轉化率上升緩慢。鄰乙基苯酚的選擇性隨TiO2負載量的增加先增大后減小,在負載量為6%時達到最高值(65.1%);2,6-二乙基苯酚的選擇性隨TiO2負載量的增加而逐漸增大;副產物苯乙醚的選擇性則隨TiO2負載量的增加而降低。綜合考慮苯酚轉化率和產物選擇性,適宜的TiO2負載量為9%。
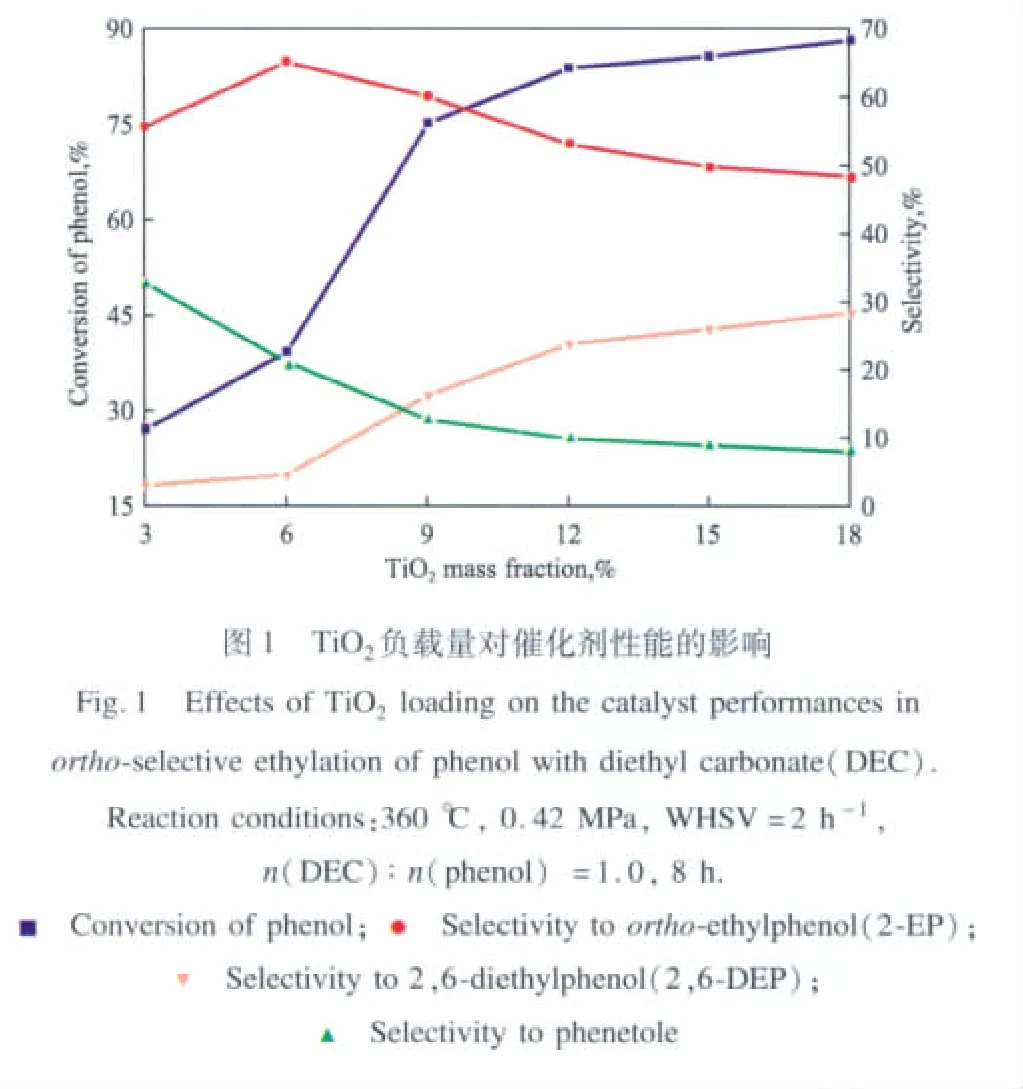
圖2為不同負載量TiO2/SiO2催化劑的XRD譜圖。由圖2可見,TiO2/SiO2催化劑的XRD譜圖與載體SiO2基本相同。當TiO2負載量為15%時,才開始出現小的銳鈦礦TiO2的特征衍射峰,而在TiO2負載量低于15%的催化劑中,沒有觀察到銳鈦礦相的存在。這一結果表明,在TiO2負載量較低時,催化劑中的TiO2高度分散在SiO2的表面;隨TiO2負載量的增加,TiO2物種開始在催化劑表面聚集,當TiO2負載量超過它在載體表面的分散容量時,便以晶體形式存在。

圖3為不同負載量TiO2/SiO2催化劑的NH3-TPD曲線。
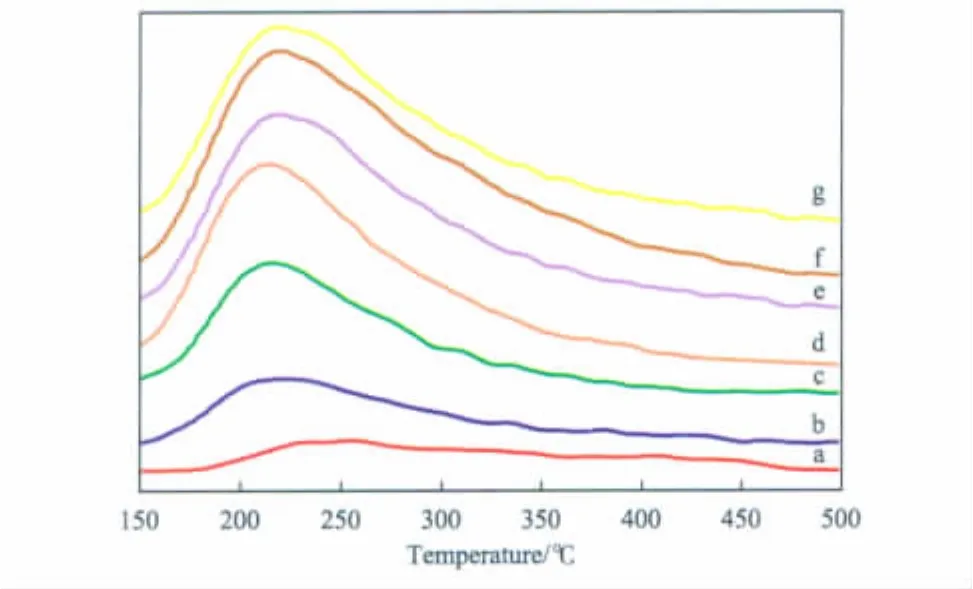
圖3 不同負載量TiO2/SiO2催化劑的NH3-TPD曲線Fig.3 NH3 -TPD curves of TiO2/SiO2catalysts with different TiO2loadings.
由圖3可見,各催化劑試樣只是在215℃附近出現了NH3脫附峰,這表明各催化劑上均只有弱酸中心,沒有強酸中心[10]。同時,隨TiO2負載量的增加,NH3脫附峰有向高溫方向偏移的傾向,但變化幅度較小,這說明負載量對催化劑的酸強度影響不大;而隨TiO2負載量的增加,弱酸中心的酸量逐漸增大。結合圖1可知,隨催化劑弱酸中心的酸量逐漸增大,苯酚的轉化率增加;當 TiO2負載量達到12%后,催化劑的酸量變化不大,苯酚的轉化率上升緩慢。由此可知,催化劑的弱酸中心有利于反應的進行,且酸量越大,催化劑的活性越高。增加TiO2負載量,催化劑中有新的弱酸中心產生,導致催化劑的酸量逐漸增大;當TiO2負載量為6%時,鄰乙基苯酚的選擇性達到最大;繼續增加TiO2的負載量,催化劑中弱酸中心的酸量繼續增大,這有利于鄰乙基苯酚和苯乙醚分別進一步發生烷基化反應,使兩者的選擇性下降。
根據文獻[11],在 Py-IR譜圖中,1 455 cm-1處的譜帶表征L酸位,而1 545 cm-1處的譜帶表征B酸位,1 490 cm-1處的吸收峰是B酸位和L酸位相互疊加的結果。圖4是不同負載量的TiO2/SiO2催化劑的Py-IR譜圖。由圖4可見,各催化劑試樣均在1 450 cm-1附近出現強的吸收譜帶,歸屬于吡啶分子與催化劑 L酸位的相互作用,而在1 545 cm-1處無吸收峰出現,這說明TiO2/SiO2催化劑只含有L酸中心,沒有B酸中心。TiO2負載量對TiO2/SiO2催化劑酸性的類型沒有明顯影響,但對催化劑的酸量有影響。結合圖4和圖1可知,隨TiO2負載量的增加,催化劑中L酸中心數量逐漸增多,苯酚的轉化率逐漸增大,這說明L酸中心是苯酚與DEC鄰位烷基化反應的活性中心。當TiO2負載量低于6%時,催化劑中的L酸中心數量較少,抑制了鄰乙基苯酚的生成;當TiO2負載量高于6%時,催化劑中的L酸中心數量明顯增多,導致苯乙醚及鄰乙基苯酚分別進一步發生烷基化反應,使得苯乙醚和鄰乙基苯酚的選擇性降低,2,6-二乙基苯酚的選擇性提高。
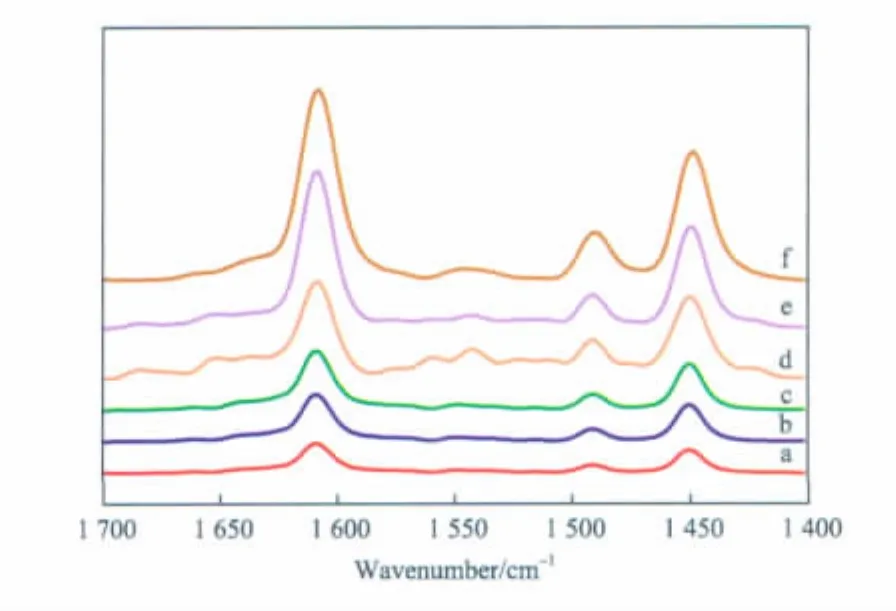
圖4 不同負載量TiO2/SiO2催化劑的Py-IR譜圖Fig.4 Py - IR spectra of TiO2/SiO2catalysts with different TiO2loadings.
2.2 反應溫度對催化劑性能的影響
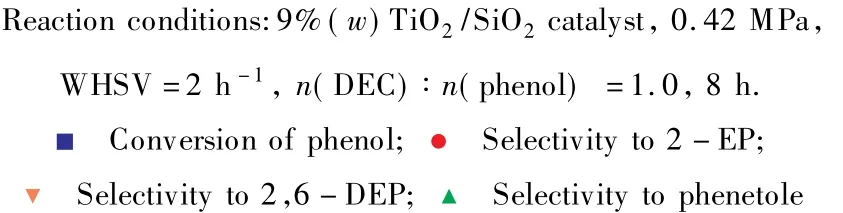
以9%TiO2/SiO2為催化劑,考察了反應溫度對苯酚與DEC鄰位烷基化反應的影響,實驗結果見圖5。由圖5可見,隨反應溫度的升高,苯酚的轉化率先增大后減小,當反應溫度為360℃時,苯酚的轉化率達到最大值(75.4%)。繼續升高反應溫度,催化劑表面會有明顯的積碳生成,導致催化劑上L酸中心數目減少[12],苯酚的轉化率出現下降趨勢。鄰乙基苯酚的選擇性受反應溫度的影響較小,幾乎保持不變。隨反應溫度的升高,2,6-二乙基苯酚的選擇性先增大后減小,副產物苯乙醚的選擇性先大幅下降后有所上升。反應溫度升高,使苯乙醚進一步發生烷基化反應的幾率增大,因此2,6-二乙基苯酚的選擇性有所增加;但反應溫度過高時,催化劑表面有明顯的積碳生成,這抑制了2,6-二乙基苯酚的生成,導致其選擇性又逐漸下降。由此可知,相對較高的反應溫度有利于提高催化劑的活性和2,6-二乙基苯酚的選擇性。綜合考慮,反應溫度選擇360℃較適宜。
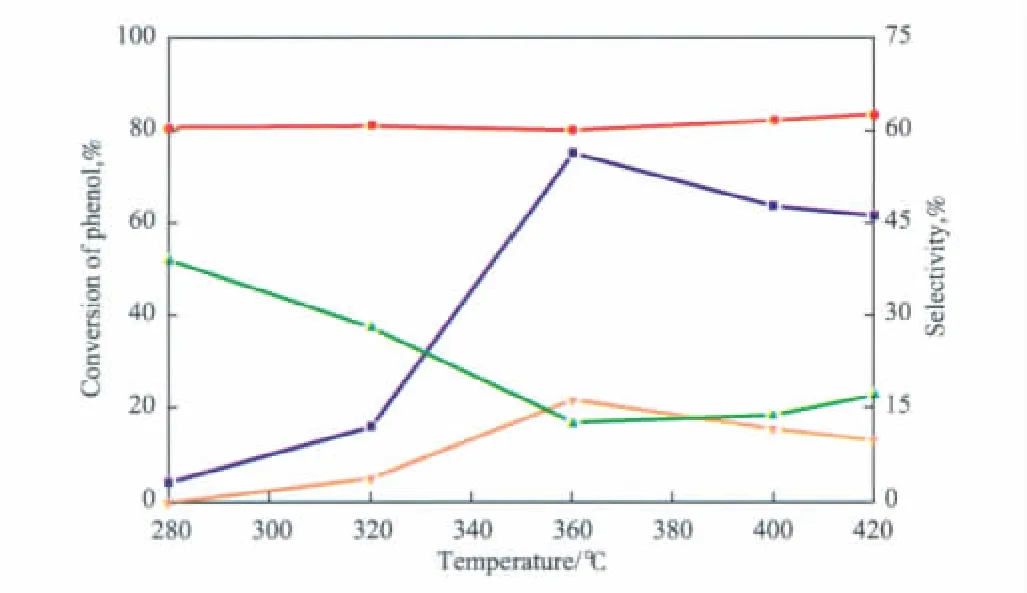
圖5 反應溫度對催化劑性能的影響Fig.5 Effect of reaction temperature on the catalyst performance.
2.3 重時空速對催化劑性能的影響
重時空速(WHSV)對苯酚與DEC鄰位烷基化反應的影響見圖6。由圖6可見,WHSV由1 h-1增至6 h-1時,苯酚的轉化率由 82.6%大幅降至36.9%;鄰乙基苯酚的選擇性明顯增加,而2,6-二乙基苯酚的選擇性卻明顯降低,副產物苯乙醚的選擇性略有增加。WHSV增大,原料與催化劑表面的接觸時間縮短,反應物分子相互碰撞發生反應的幾率減小,烷基化反應進行的不充分,導致苯酚的轉化率大幅下降。但WHSV較小時,反應物分子在催化劑表面停留時間過長,增加了鄰乙基苯酚及苯乙醚分別進一步發生烷基化反應的幾率,因此這兩者的選擇性均隨WHSV的增大而增加。根據產物選擇性隨轉化率變化的趨勢可以推測,2,6-二乙基苯酚主要是由鄰乙基苯酚及苯乙醚分別進一步反應獲得的[13-14]。綜合考慮苯酚轉化率和產物選擇性,WHSV選擇2 h-1較為適宜。
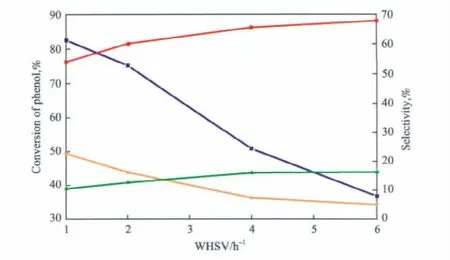
圖6 WHSV對催化劑性能的影響Fig.6 Effect of WHSV on the catalyst performance.
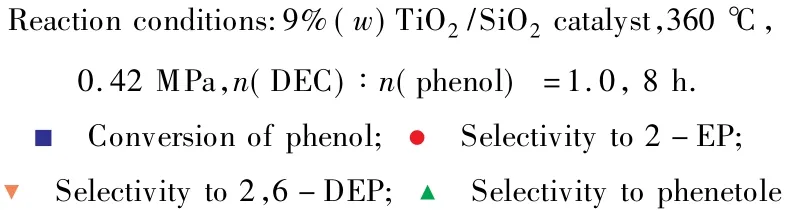
2.4 原料配比對催化劑性能的影響
n(DEC)∶n(苯酚)對苯酚與DEC鄰位烷基化反應的影響見圖7。
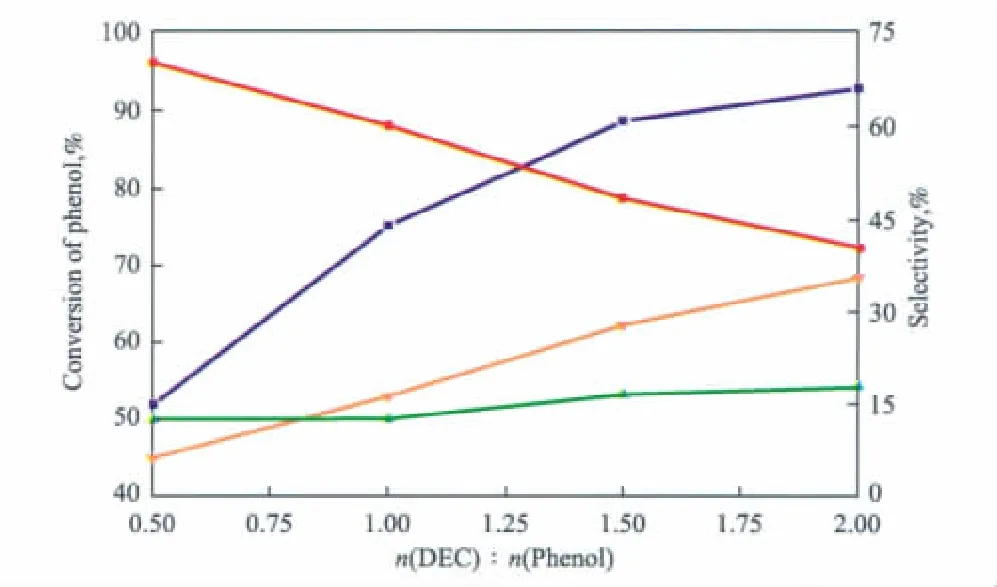
圖7 n(DEC)∶n(苯酚)對催化劑性能的影響Fig.7 Effect of n(DEC) ∶n(phenol)on the catalyst performance.
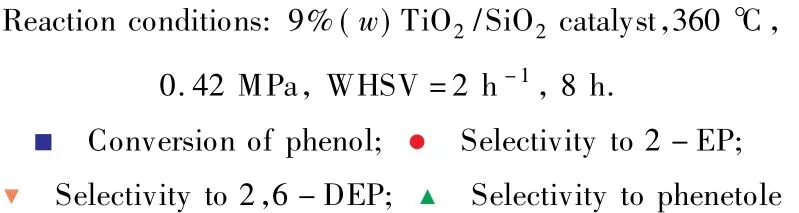
由圖7可見,隨n(DEC)∶n(苯酚)的增大,即原料中DEC含量的增加,苯酚的轉化率增大,當n(DEC)∶n(苯酚)=2.0時,苯酚的轉化率為92.9%;鄰乙基苯酚的選擇性逐漸減小,2,6-二乙基苯酚的選擇性卻明顯增加,副產物苯乙醚的選擇性略有增加。隨原料中DEC含量的增加,在催化劑表面解離出來的乙基自由基增多,與苯酚分子發生碰撞的幾率增大,因此苯酚的轉化率增加[15]。但隨催化劑表面解離出來的乙基自由基的增多,鄰乙基苯酚與乙基自由基進一步發生烷基化反應生成2,6-二乙基苯酚的幾率也增大,導致鄰乙基苯酚的選擇性逐漸減小,2,6-二乙基苯酚的選擇性明顯增加。綜合考慮苯酚轉化率和產物選擇性,選擇n(DEC)∶n(苯酚)=1.0 較為適宜。
3 結論
(1)TiO2/SiO2催化劑中,L酸中心是苯酚與DEC鄰位烷基化反應的主要活性中心;TiO2負載到SiO2上可產生較強的弱酸中心,隨TiO2負載量的增加,酸量逐漸增加,催化劑的活性逐漸增強;較多的L酸中心有利于生成的鄰乙基苯酚進一步轉化為2,6-二乙基苯酚。
(2)以9%TiO2/SiO2為催化劑,苯酚與DEC鄰位烷基化的適宜反應條件為:360℃,0.42 MPa,n(DEC)∶n(苯酚)=1.0,WHSV=2 h-1,8 h。在此條件下,苯酚的轉化率為75.4%,鄰乙基苯酚和2,6-二乙基苯酚的選擇性分別為60.2%和16.3%。
[1]Vinu A,Karthik M,Miyahara M,et al.ortho-Selective Ethylation of Phenol with Ethanol Catalyzed by Bimetallic Mesoporous Catalyst,CoAl- MCM -41[J].J Mol Catal A:Chem,2005,203(1 -2):151-157.
[2]Shanmugapriya K,Anuradha R,Palanichamy M,et al.Vapour Phase Reaction of Phenol with Ethyl Acetate over MCM-41 Molecular Sieves[J].J Mol Catal A:Chem,2004,221(1 -2):145-153.
[3]Mathew T,Shiju N R,Bokade W,et al.Selective Catalytic Synthesis of 2-Ethyl Phenol over Cu1-xCoxFe2O4-Kinetics,Catalysis and XPS Aspects[J].Catal Lett,2004,94(3 - 4):223-236.
[4]石勤智,史建公,曹鋼.β-沸石催化合成乙基苯酚的工藝研究[J].精細石油化工,2009,26(5):35-38.
[5]馬新賓,張震,石海峰,等.碳酸二乙酯的合成方法[J].化學通報,2003,66(8):528-535.
[6]潘鶴林,田恒水,朱云峰.碳酸二乙酯合成方法綜述[J].上海化工,2001,(20):23-26.
[7]Li Yongxin,Xue Bing,He Xueyi.Synthesis of Ethylbenzene by Alkylation of Benzene with Diethyl Carbonate over Parent MCM-22 and Hydrothermally Treated MCM -22[J].J Mol Catal A:Chem,2009,301(1-2):106-113.
[8]劉秀梅,郭新聞,劉民.離子液體催化苯酚與叔丁醇烷基化反應[J].石油學報,2008,24(2):216-221.
[9]李杰,劉杰.苯酚烷基化催化劑研究進展[J].應用化工,2004,(4):11 -13.
[10]Lonyi F,Valyon J.On the Interpretation of the NH3-TPD Patterns of H-ZSM -5 and H -Mordenite[J].Microporous Mesoporous Mater,2001,47(2-3):293-301.
[11]Hughes T R,White H M.A Study of Surface Structure of Decationized Y Zeolite by Quantitative Infrared Spectroscopy[J].J Phys Chem,1967,71(7):2192 -2201.
[12]Udayakumar S,Pandurangan A,Sinha P K.Para-Selective Ethylation of Phenol with Diethyl Carbonate over Mesoporous Al-MCM -41 Molecular Sieves[J].Appl Catal,A,2004,272(1 -2):267 -279.
[13]Gandhe A R,Fernandes J B.A Highlyortho-Selective TiO2Catalyst for the Methylation of Phenol[J].Catal Commun,2004,5(2):89 -94.
[14]徐磊,吳淑杰,張文祥,等.鐵鋯氧化物催化劑上苯酚和甲醇氣相鄰位烷基化反應[J].物理化學學報,2009,25(2):242-246.
[15]李永昕,張丹慧,薛冰.碳酸二乙酯與苯酚催化合成苯乙醚[J].石油化工,2007,36(11):1157 -1161.
ortho-Selective Ethylation of Phenol with Diethyl Carbonate over TiO2/SiO2Catalyst
Lü Liechao,Xue Bing,Xu Jie,Li Yongxin
(Institute of Petrochemical Technology,Changzhou University,Changzhou Jiangsu 213164,China)
TiO2/SiO2catalysts were prepared by an impregnation method and characterized by means of XRD,NH3-TPD and Py-IR.ortho-Selective ethylation of phenol with diethyl carbonate(DEC)was carried out over the prepared TiO2/SiO2catalysts in a continuous flow fixed-bed reactor.The effects of TiO2loading and reaction conditions on the catalyst performances were investigated.The results indicated that the Lewis acid sites on the TiO2/SiO2catalysts led to high activity of the catalysts.With increase of TiO2loading,the conversion of phenol and the selectivity to 2,6 -diethylphenol increased,and the selectivity toortho-ethylphenol first rose and then lowered when TiO2loading reached 9%(w).Under the optimal reaction conditions of 9%(w)TiO2/SiO2as the catalyst,360 ℃,0.42 MPa,n(DEC) ∶n(phenol)1.0,WHSV 2 h-1and 8 h,the phenol conversion was 75.4%,and the selectivities toortho-ethylphenol and 2,6 -diethylphenol were 60.2%and 16.3%respectively.
titania/silica catalyst;diethyl carbonate;phenol;ortho-ethylphenol;2,6-diethylphenol
1000-8144(2011)04-0376-05
TQ 032.4
A
2010-10-28;[修改稿日期]2011-01-30。
呂列超(1984—),男,江蘇省新沂市人,碩士生,電郵dachao609@163.com。聯系人:李永昕,電話 0519-86330135,電郵liyx@cczu.edu.cn。
江蘇省高技術研究項目(BG2006015)。
(編輯 安 靜)
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
