海水電池用鎂陽極的研究與應用
2011-11-03 03:32:18王日初彭超群
中國有色金屬學報 2011年2期
馮 艷,王日初,彭超群
(中南大學 材料科學與工程學院,長沙 410083)
海水電池用鎂陽極的研究與應用
馮 艷,王日初,彭超群
(中南大學 材料科學與工程學院,長沙 410083)
鎂陽極材料具有電負性好,比能量高以及密度小等優異性能,在海水電池領域具有廣闊的應用前景。綜述了近年來國內外鎂陽極海水電池的研究及應用,討論鎂陽極材料的活化機制和腐蝕行為,探討合金元素和第二相對鎂陽極電化學活性和耐腐蝕性能的影響,指出今后海水電池用鎂陽極材料的發展應在充分研究合金元素活化機理和第二相影響的基礎上,研制出自腐蝕速率更小、陽極利用率更高以及比能量更大的新型鎂合金陽極材料。關鍵詞:海水電池;鎂陽極;活化;腐蝕;合金元素;第二相
海水電池是在第二次世界戰爭期間由美國貝爾實驗室設計、通用電氣公司研制的,它依靠陽極金屬材料在海水中的腐蝕溶解提供陽極放電電流,而陰極則主要依靠海水中的溶解氧在惰性的氣體電極上進行還原反應提供陰極電流。海水電池最突出的特點是不需要攜帶電解質,可以在需要的時候利用天然海水形成電解液。出于不同使用目的,海水電池具有多種不同類型,如大功率水下武器裝備的動力電池,長周期、小功率的水中探測儀器類電池以及水下航行體的動力電池——半燃料海水電池等[1]。其中大功率動力電池的應用前景最廣,技術難度最大,研制也最有戰略意義,目前制約其性能的重要一方面就在陽極材料的開發上[2]。自從20世紀40年代以來,美國和一些發達國家的政府和商業機構就已經開始研究和研制在海水中的大功率動力電池[3?5]。
成功應用在大功率海水電池中的陽極材料集中在鎂合金和鋁合金上[3?7],這主要是由于鎂、鋁具有優于其他金屬的陽極性能,如電極電位負、密度小、比容量高等。常用的陽極金屬鋰電位最負,為?3.05 V,但化學性質過于活潑,無法用于水溶液類電解液電池,至今鋰由于安全性差而無法應用于大功率放電。事實上,鎂、鋁自身也有作為陽極材料不可回避的缺陷。鋁比能量高于鎂的,但其陽極電位在同等條件下低于鎂的,應用于海水電池時其反應產物為絮狀沉淀物Al(OH)3,易造成腐蝕產物堆積而影響電池性能,同時由于鋁是兩性金屬,容易與介質發生嚴重析氫反應[7]。鎂具有較高的電化學活性,陽極電位為?2.363 V,電化學當量為2 200 A·h/kg,僅低于鋰和鋁的[8]。純鎂表面的微觀腐蝕電池驅動力大,易發生微觀腐蝕原電池反應,產生大量氫氣,導致陽極的法拉第效率降低。20世紀60~80年代,國外已對鎂陽極進行了廣泛的研究與試驗,普遍采用合金化的方法對鎂陽極進行性能優化。研究結果表明,通過合金化的鎂合金能夠在較大電流密度下減小自放電,得到60%的效率,比鋁合金更適合作為冷的海水溶氧電池的陽極[1]。目前已開發的應用于大功率海水電池的鎂陽極材料有英國鎂電子公司生產的AP65和MT75以及俄羅斯和國內中南大學研制的Mg-Hg陽極材料[8],具體成分和制備工藝未見報道。
鎂合金陽極雖然在大功率海水電池中得到應用,但現有的鎂合金陽極材料仍存在自腐蝕速度大、陽極利用率低等不足之處,尋找陽極利用率高的鎂合金陽極材料是國際上鎂電池研究的熱點和難點問題。對鎂陽極的合金化元素的設計需要人們對合金元素作用的認識進一步提高,如Zn、Bi、Pb和In等,其活化機理仍沒有統一的解釋[9]。本文作者從活化機制、腐蝕行為、合金元素以及第二相對海水電池用鎂陽極性能的影響等方面,探討海水電池用鎂陽極的發展方向。
1 常用鎂陽極海水電池
1.1 鎂/氯化銀海水電池
鎂/氯化銀海水電池采用金屬 Mg做陽極,AgCl做陰極,反應原理如下[1]:
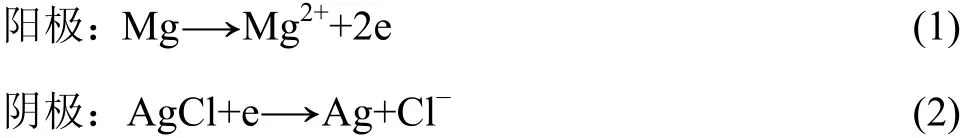
鎂陽極在海水中能長期保持其活性,因為氯化物海水是鎂陽極很好的活化溶液,同時由于鎂的極化較大,因此電極反應的熱效應較大,這一熱量保證了該電池具有良好的低溫性能,無需輔助加熱裝置就可適應?60 ℃低溫;選用溶解度低的 AgCl作為陽極,AgCl/Ag電位非常穩定,能作為中性溶液中的參比電極使用,其放電后轉化為導電性良好的 Ag,減少Mg/AgCl海水電池內阻,使其適宜于大電流密度下工作,比能量可達88 W·h/kg[10?11]。由于靠海水激活,因此Mg/AgCl海水電池平時處于干態保存,擱置時間可長達5 a,但這一體系需要消耗貴金屬Ag,造價高,且總功率有待提高。
Mg/AgCl海水電池作為一次激活貯備電池,采用雙極性堆式結構[12]。電池的陽極為含少量 Al、Zn和Pb等元素的合金,與純Mg電池相比,其比能量提高20%。這種電池20世紀60年代開始在美國的MK44魚雷、MK45魚雷上使用,迄今已有40多年的歷史。目前正在服役的意大利A244 /S魚雷、英國的“鯆魚”魚雷、法國的 R3魚雷都以這種電池為動力[13],但電池結構不同,性能各異,其性能如表1所列[14?15]。
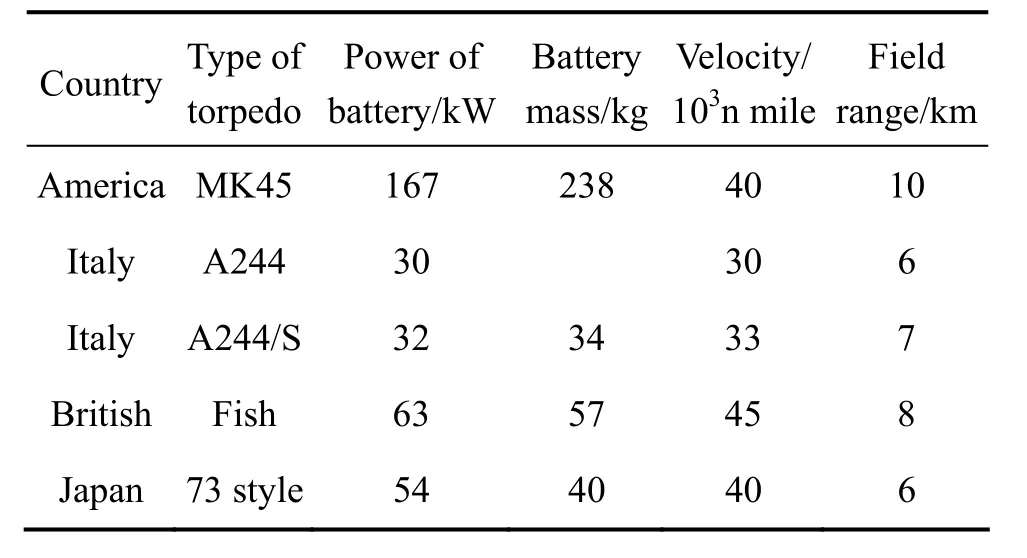
表1 各國魚雷動力電池性能[14?15]Table 1 Performances of battery for torpedo in many countries[14?15]
1.2 鎂/氯化亞銅海水電池
這是由前蘇聯開發的海水電池,目前俄羅斯仍然大量使用。該電池的陰極材料采用相對經濟的銅合金材料代替貴金屬銀。為防止其氧化,加入一定數量的SnCl2,同時采用氬氣保護措施,保證電極的活性。采用鎂汞合金作陽極,汞齊化主要是提高鎂的穩定性和表面的析氫過電位,抑制鎂陽極的自腐蝕。鎂/氯化亞銅海水電池電極反應原理如下:
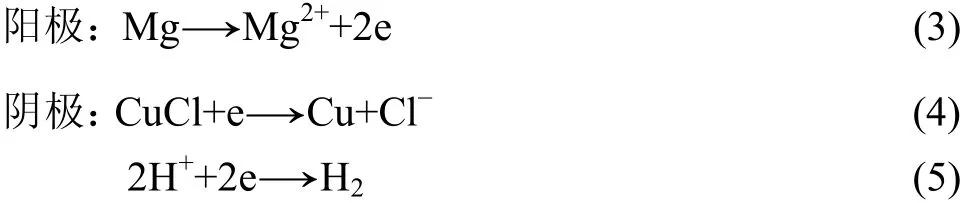
該電池造價只有Al/AgO海水電池的1/3,但電池體積較大,目前只有俄羅斯實際應用到 y ?T T 、TT?80型魚雷上。王日初等[16]也對這種電池的陽極材料做了大量研究,開發了Mg-Hg-Ga陽極材料。研究表明,這種陽極材料在 200 mA/cm2的電流密度下,平均電壓達到?1.8 V,陽極極化小,具有很好的電化學活性。
2 海水電池用鎂陽極的活化機理
迄今為止,對鎂陽極活化機理的研究還大多數集中在二元和三元合金上,而且沒有得出統一觀點,然而現在鎂合金陽極已經發展到五元甚至七元[17],其反應機理也變得更加復雜。因此,在研究鎂合金陽極活化機理時,鎂合金的微觀組織、鎂合金中各元素的相互作用以及合金元素與海洋環境中 Cl?電解液之間的相互作用是都鎂合金陽極活化的關鍵。
馮艷等[16]提出含Hg、Ga元素的鎂陽極活化機理:溶解—再沉積。他們認為:在溶解初期,陰極性第二相化合物通過腐蝕原電池反應引發鎂基體點蝕的出現,促進了Mg基體和合金元素Hg、Ga的溶解;在放電過程中,溶液中的Hg+和Ga3+被Mg還原成Hg、Ga而沉積在陽極表面,反應原理如下:

Hg和Ga沉積層一方面形成鎂汞齊,鎂汞齊與水發生劇烈反應而繼續產生Hg和Ga沉積,此循環促進活化,反應式如下:

Hg,Ga沉積層另一方面影響鎂陽極表面結構(見圖1),隔離腐蝕產物,破壞氧化物膜,從而使合金電位負移,點蝕更易引發和擴散,進一步起到活化作用。

圖1 Mg-Hg-Ga陽極放電過程的表面結構示意圖Fig.1 Schematic diagram of surface structure of Mg-Hg-Ga anode during electric
3 海水電池用鎂陽極的腐蝕
3.1 腐蝕類型
從腐蝕形貌來看,鎂陽極的腐蝕并不是均勻腐蝕,其不均勻程度與合金的種類及腐蝕環境有關[18],海洋環境中的 Cl?使鎂陽極局部區域置于酸性環境中,加速腐蝕[19?20]。在很多情況下,鎂陽極的腐蝕一般都是從某一局部腐蝕開始的,常常表現為點蝕,其腐蝕小孔可深可淺,然后發展開來。一般來說,點蝕、電偶腐蝕和晶間腐蝕是鎂陽極材料中最常見和最重要的腐蝕類型[21?22]。
3.1.1 點蝕
在鎂合金陽極表面有一層氧化膜,氧化膜的致密度為氧化物分子體積與形成該氧化物的金屬原子體積之比[23],鎂的致密度小于 1,說明其氧化物膜疏松、多孔,存在缺陷,容易造成點蝕。另外,第二相化合物和基體之間的微電池反應也是產生點蝕的原因之一。在微電池反應中,第二相為陰極,基體為陽極,在第二相周圍的基體發生溶解,最后導致第二相脫落,從而產生點蝕坑[24]。圖2所示為Mg-Hg-Ga陽極材料蝕坑的截面形貌[8]。

圖2 Mg-Hg-Ga合金在NaCl溶液中蝕坑的截面形貌[8]Fig.2 Morphology of cross-section of pit in Mg-Hg-Ga alloys in NaCl solution[8]
圖3 所示為鎂合金表面點蝕發展示意圖。以此說明點蝕發展的過程[8]。當點蝕發生后,點蝕坑底部金屬鎂便發生溶解,即 Mg→Mg2++2e,孔內金屬離子不斷增加。在含有氯離子的水溶液中,在蝕孔電池產生的電場作用下,蝕孔外陰離子(Cl-)不斷向孔內遷移、富集,孔內氯離子濃度升高,同時由于孔內金屬離子濃度升高并發生水解:Mg2++2H2O→Mg(OH)2+2H+,使孔內溶液氫離子濃度升高,pH值降低,溶液酸化,相當于使蝕孔內金屬處于HCl介質中,處于活化溶解狀態。水解產生的氫離子和孔內的氯離子又促使蝕孔側壁的鎂繼續溶解,發生自催化反應:Mg+2H+→Mg2++H2。由于孔內濃鹽溶液中氧的溶解度很低,又加上擴散困難,使得閉塞電池局部供氧受到限制,孔內的鎂難以再形成氧化膜,而一直處于活化狀態。蝕孔口形成的Mg(OH)2腐蝕產物沉積層,阻礙了擴散和對流,使孔內溶液得不到稀釋,從而造成上述電池效應。
3.1.2 電偶腐蝕
鎂陽極中不僅存在不同的相、雜質與缺陷,即使是同一基體相,其不同部位合金元素的含量也不同,故鎂合金表面是不可能電化學均勻的,因此鎂陽極易由于上述微觀結構特征引起局部電偶腐蝕。具體來說,鎂陽極中引起腐蝕微電偶的原因有以下幾種。
1) 成分不均勻
在鎂陽極基體相中,成分的分布并不均勻,如初生的基體相與共晶的基體相或包晶的基體相在合金元素的含量上就有很大不同。這些成分上的不同,導致鎂陽極表面電化學行為不同,因此,鎂陽極在基體相中會形成電偶腐蝕。
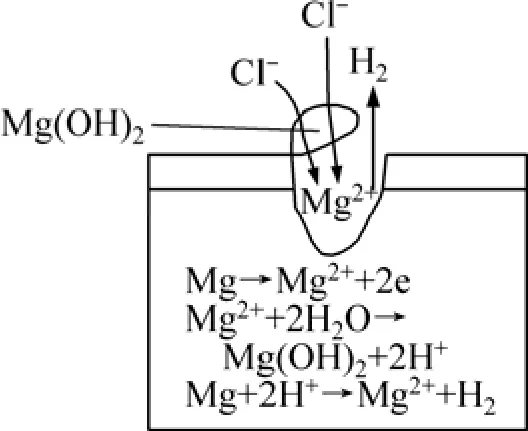
圖3 鎂合金點蝕發展示意圖[8]Fig.3 Schematic diagram of pitting development of Mg alloys[8]

圖4 Mg-Hg-Ga合金時效不同時間后第二相的形貌Fig.4 Morphologies of second-phases in Mg-Hg-Ga alloy after aging for different times: (a) 1 h; (b) 200 h
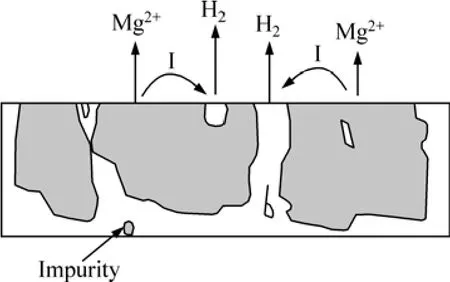
圖5 雜質元素引起鎂合金的電偶腐蝕示意圖[28]Fig.5 Schematic diagram of electric corrosion of Mg alloy due to impurity[28]
2) 第二相
為了獲得更高電化學活性和電流利用率的鎂陽極,一般在鎂中添加各種合金元素形成第二相[25],如Mg-Hg-Ga合金中的Mg3Hg和Mg5Ga2,Mg-Pb合金中的Mg2Pb,Mg-Sn合金中的Mg2Sn,Mg-Sr合金中的Mg9Sr,Mg-Zn合金中的Mg2Zn等。鎂陽極中的這些第二相,其自腐蝕電位一般都比基體相高。有人定義了一個驅動力的概念來描述合金元素相對鎂腐蝕速率的影響[26],即在鎂合金中,某一相的腐蝕驅動力應該等于該相的電位與鎂的電位差,再減去該相的析氫過電位。雖然這是一個極其粗略的評估第二相對基體相電偶效應大小的方法,但它實際上已經考慮了第二相對基體相電偶效應的主要因素。
事實上,第二相對鎂基體的腐蝕極大取決于鎂陽極合金中第二相的數量和分布。以Mg-Hg-Ga為例,圖4所示為同一成分的Mg-Hg-Ga合金時效不同時間后的微觀形貌[27],EDS和XRD分析顯示,圖4(a)中Mg3Hg和α-Mg形成共晶組織,呈網狀分布在晶界,而圖4(b)中的第二相Mg3Hg則成塊狀均勻彌散地分布在整個晶體中。由這兩個合金的動電位極化曲線掃描結果可知,當第二相Mg3Hg與基相α-Mg形成共晶在晶界呈網狀分布時其腐蝕電流密度為70.92 mA/cm2,而第二相 Mg3Hg在晶內呈塊狀均勻彌散分布時其腐蝕電流密度為 2.34 mA/cm2,比網狀共晶分布時小很多。這說明陰極性第二相Mg3Hg均勻彌散分布時,可對基體 α-Mg的腐蝕起到阻礙作用,提高 Mg-Hg-Ga陽極的耐腐蝕性能[27]。
3) 雜質元素
一般來說,雜質元素及其化合物具有比鎂基體相正得多的電極電位,而且,這些雜質的陰極析氫過電位都很低,與鎂合金構成微電池時,它們都是十分有效的陰極相,能極大地促進鎂合金的電偶腐蝕。這是雜質對鎂合金腐蝕的直接加速作用,其腐蝕作用示意圖如圖5所示[28]。
固溶于鎂合金中的雜質雖然對鎂合金的腐蝕影響不大,但可能隨鎂合金的腐蝕而溶解到溶液中,而后再被鎂還原成單質沉積回鎂的表面形成腐蝕電偶,促進其沉積處鎂合金的進一步腐蝕。這種不利作用是雜質的二次影響。
4) 由局部腐蝕引發的電偶腐蝕
不僅上述的這些鎂陽極本身的微觀組織形成的微電偶導致電偶腐蝕的發生,鎂陽極中局部腐蝕的發生本身也是一種腐蝕微電偶。例如,腐蝕過程中先發生的點蝕導致表面膜破裂,這些破裂處的表面與表面膜未破裂處的金屬存在電化學差異,從而導致電偶腐蝕發生,使得鎂陽極表面的腐蝕進一步擴展。
3.1.3 晶間腐蝕
晶間腐蝕是指腐蝕沿著金屬或合金的晶粒邊界或它的鄰近區域發展;而晶粒本身的腐蝕是很輕微的一種腐蝕類型,它是由晶粒與晶界之間的電位差引起的局部腐蝕。鎂合金陽極由于其較高的電化學活性,置于海洋性大氣環境等腐蝕性強的介質中時,有時會發生晶間腐蝕。這種腐蝕使晶粒間結合力大大降低,降低了鎂陽極材料的電流效率。
含有不同合金元素的鎂陽極,其晶間腐蝕的傾向不同。例如含鋁的鎂陽極,由于鋁在晶粒內部含量低于晶粒周邊的,一般是晶粒內部先被腐蝕,其發生晶間腐蝕的傾向很小,但若Mg、Al形成大量的MgAl、Mg2Al3、Mg4Al3等金屬間化合物[25],這些金屬間化合物在晶間的存在,會引起晶間腐蝕,增大鎂的自腐蝕速度,加速固溶體的破壞;含鋯的鎂陽極晶界上的第二相是相對穩定的,合金元素鋯主要分布于晶內而發生晶間腐蝕;Mg-1%Ga(質量分數)合金由于晶界生成Mg5Ga2相,腐蝕沿著晶粒邊界或它的鄰近區域發生,而晶粒本身腐蝕很輕微,圖6所示為Mg-1%Ga合金的晶界腐蝕形貌。

圖6 Mg-1%Ga合金的晶界腐蝕形貌Fig.6 Morphology of grain-boundary corrosion in Mg-1%Ga alloy
3.2 腐蝕機理
3.2.1 總腐蝕反應
鎂合金的腐蝕性由合金中單個組元相的腐蝕反應控制,如果合金中含有與環境反應激烈的組元,則耐腐蝕性能很差。純鎂的腐蝕反應很重要,提供理解各種鎂合金腐蝕的基礎。
鎂在水溶液環境中溶解是發生與水的電化學反應,這個反應產生氫氧化鎂和氫氣,因此鎂的腐蝕對氧氣的濃度不敏感[29?31],然而氧氣的存在在空氣腐蝕中是很重要的因素[32]。鎂在水溶液環境中的腐蝕通常包括陰陽兩極之間的微電偶腐蝕[33]。

陽極反應式(10)還包括產生存在時間不長的單價鎂離子(Mg+)的中間步驟[30,34]。氫離子的還原過程和陰極相的析氫過電位在鎂的腐蝕中起重要作用,低析氫過電位的陰極相使析氫便利,產生較大的腐蝕速率[32]。
目前,關于鎂合金腐蝕反應的系統研究還沒有報道。有研究表明,鎂合金的腐蝕反應類似于純鎂的反應。例如,SONG等[35?36]研究了 Mg-Al-Zn合金的陽極溶解,結果表明Mg是溶液中溶解的主要組元,Al發生少量溶解,沒有發現 Zn在溶液中溶解。這意味著對于Mg-Al-Zn合金來說,反應式(9)~(12)的4個反應可以反映其主要的腐蝕過程,當然,也不能排除合金元素Al、Zn對鎂腐蝕反應速率的作用。
3.2.2 負差數效應
一般而言,鐵、銅合金在酸性溶液中隨著極化電位的升高,其陽極溶解率增加,陰極析氫速率降低,但鎂合金的析氫腐蝕反而增加。這種行為看似與電化學原理相沖突,被定義為負差數效應,見圖 7[37]。從曲線A-B-F-C可知,極化電位從負變到正值時,鎂合金的析氫速率開始降低,然后升高。
SONG等[28]根據對實驗數據的仔細分析,提出了一種新的“陽極析氫”的腐蝕機制來解釋負差數效應,見圖8。
鎂合金的表面都存在一層白色的 Mg(OH)2保護膜層,但是這層膜并不完整致密,在鎂合金服役期間,這層膜的不完整性隨著極化電位的升高而加大,正是這層表面膜的破裂對鎂合金的腐蝕起著決定性的作用。
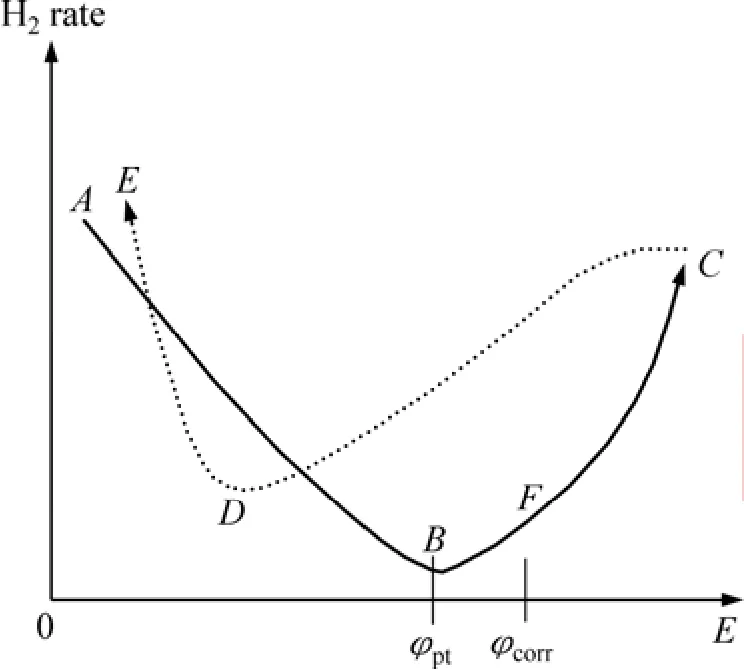
圖7 鎂合金析氫速率與極化電位關系示意圖[37]Fig.7 Schematic diagram of dependence of hydrogen evolution rate from polarization magnesium alloy[37]
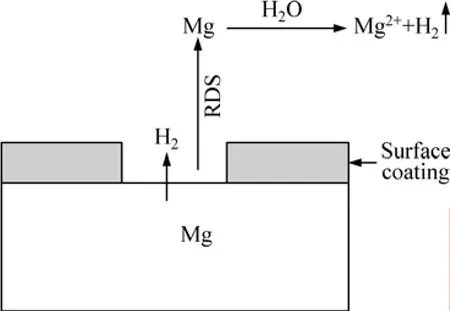
圖8 鎂腐蝕機理示意圖[37?40]Fig.8 Schematic diagram of corrosion mechanism of magnesium[37?40]
當電壓低于點蝕電位時,鎂合金整個表面覆蓋完整的保護膜并且合金的腐蝕速率很低。此時主要是陰極析氫反應:

且析氫速率隨著極化電壓的升高而降低,直到到達鎂合金的點蝕電位。
到達點蝕電位時,鎂合金表面膜由于點蝕的作用開始脫落,在鎂合金基體裸露區,下列產生單價鎂的腐蝕反應發生:

單價的鎂離子與水反應生成更為穩定的二價鎂的腐蝕產物,此時陽極析氫反應也隨之發生:
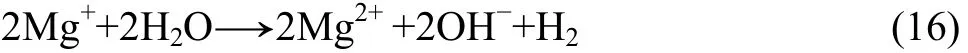
隨著電極電位的正移,鎂陽極溶解與“陽極析氫”的速度增大,同時由于表面膜的破壞更嚴重,裸露的金屬面積增大,更多鎂基體的溶解也使陰極析氫更容易。因此總的析氫速度(為陽極析氫與陰極析氫的總和)就會隨著電位的正移而增大,形成負差數效應,而且在腐蝕后期,由于嚴重的局部或不均勻腐蝕,導致一些未腐蝕的鎂顆粒脫落,負差數效應更明顯。解釋鎂合金負差數效應的腐蝕模型的要點可總結為以下幾個階段:
1) 鎂陽極材料具有很負的點蝕電位(見圖 7),當極化電位達到點蝕電位并繼續正移,導致鎂合金表面保護膜的破壞;
2) 單價鎂離子的溶解以及膜破壞處的陰陽極析氫反應(反應式(13)~(15))使得總的析氫腐蝕速率增加;
3) 腐蝕后期,嚴重的局部或不均勻腐蝕導致鎂顆粒的不均勻脫落。
在此基礎上,LIU等[41]根據 Tafel動力學公式對鎂陽極的負差數效應用數學模型進行了解釋。
4 合金元素和第二相對鎂陽極電化學性能的影響
4.1 合金元素
理論上純鎂的電極電位很負,為?2.363 V,但實際應用中,純鎂在水中會產生保護性強的氫氧化鎂膜,使材料快速極化[42],因此純鎂不適合做大功率海水電池用陽極。化學成分,即合金元素的種類和含量是影響鎂陽極性能最主要的因素。在已有的鎂合金陽極中,常用的活化元素主要是Hg、Ga、Sn、Pb、Zn、Mn、Al和Tl等,這些元素都在一定程度上使鎂合金的電位負移,改善陽極的電化學活性。
1) 合金元素Hg
研究表明,Hg能通過溶解-再沉積破壞鎂表面的鈍化膜,維持陽極材料的活化溶解[43]。合金元素 Hg還能有效地抑制鎂合金的析氫腐蝕,這與其具有較高的析氫過電位有關。但當Hg的添加量過高時,Hg與Mg形成第二相,易在晶界處析出共晶組織,加速鎂陽極的自腐蝕。
2) 合金元素Ga
Ga能與其他合金元素,如Hg、Sn、Pb等,在電極工作溫度(60~100 ℃)下形成低共熔混合物,破壞鎂表面鈍化膜。在含Hg的電解液中,由于Hg和Ga的共同沉積(沉積在鎂氧化膜缺陷部位),對鎂陽極產生活化作用。必須指出的是,隨著 Ga含量的增加,鎂合金陽極的電位變負,但當添加量過高時,Ga與Mg形成第二相,在晶界處析出共晶組織,將明顯降低電流效率[44]。
3) 合金元素Sn
合金元素 Sn具有較高的析氫過電位,能有效地抑制析氫腐蝕,并能與Ga、In等其他合金元素形成低共熔混合物,破壞Mg表面的鈍化膜。Sn還能與Mg形成Mg2Sn相,有利于鈍化膜的破裂,使電極反應深入,但同時降低鎂合金的耐腐蝕性能[8]。
4) 合金元素Pb
合金元素Pb能與Ga元素形成低共熔混合物,破壞鎂表面的鈍化膜。Pb的電極電位較Mg正,在電解液中形成微觀腐蝕原電池,使鎂陽極的電位向正方向漂移。Pb與Al一起加入,降低鎂的加工性能,但可改善鎂合金陽極的有效工作電位和陽極的電流效率,如AP65合金[45]。
5) 合金元素Zn
Zn 和其他合金元素(Sn、In、Hg、Bi)[45?46]等一起能有效地降低純鎂表面氧化膜的穩定性,從而獲得陽極的高活性。Zn能強化鎂基體,但對鑄造性能有不利影響,有形成疏松和熱裂紋的傾向。Zn還可提高含Cu、Ni雜質的鎂陽極的穩定性,有利于提高陽極的電流效率,但過高則反而會使電流效率降低。一般而言,Zn的質量分數應低于4%[47]。
6) 合金元素Mn
Mn在鎂中的溶解度為3.4%,如果熔煉方法控制得當,可得到含有少量Mn晶體的單相固溶體組織。Mn在鎂中主要是凈化合金組織,消除雜質元素Fe的影響,減小腐蝕速率,從而提高電流效率。Mn的另一個作用是使 Mg-Mn陽極表面形成比氫氧化鎂膜更具有保護作用的水化二氧化錳膜,使析氫作用進一步減弱[48]。最近,有人將少量Ca添加到Mg-Mn合金中,Ca使Mg-Mn合金晶粒細化,并在鎂基體的晶界上析出Mg2Ca陰極性化合物,使得晶間腐蝕傾向降低,減少晶粒的脫落,從而提高電流效率,達到62.36%,而且驅動電壓也有所增大[48?49]。
7) 合金元素Al
Al元素的加入能使鎂陽極表面生成可導電的氫氧化物膜以及細顆粒的反應產物,并迅速從鎂陽極表面剝落,有助于提高鎂陽極的電流效率[50]。控制 Al的最大含量也至關重要,因為過量的Al容易與Mg反應生成具有陰極特性的中間產物Mg2Al3,這種中間產物會導致晶間腐蝕的發生,降低電流效率,因此鎂陽極中的Al含量不宜過高。Al還能提高鎂合金強度[47]。早期使用的 Mg陽極材料以及現今使用的大部分 Mg犧牲陽極材料均采用 AZ系列的鎂合金,如 AZ31、AZ61和AZ91。
8) 合金元素Tl
Tl元素與Hg、Pb等元素一樣屬于具有高析氫過電位的重金屬元素,Tl能破壞鎂陽極表面的保護膜,減少腐蝕產物成泥,降低陽極極化,提高工作電位。英國國防部開發使用的MTA 75(Mg-7%Tl-5%Al)陽極具有比AP65合金更高的工作電位[51]。
4.2 第二相
對于不同的陽極材料,必須考慮第二相粒子的類別、特性等對其電化學性能的影響。馮艷等[16]研究了不同的第二相粒子 Mg3Hg、Mg5Ga2、Mg21Ga5Hg3對Mg-Hg-Ga陽極材料電化學性能的影響,指出這3個第二相對Mg-Hg-Ga陽極的電化學活性的促進作用從高到低依次為Mg3Hg、Mg21Ga5Hg3、Mg5Ga2,交流阻抗分析表明第二相Mg3Hg使Mg-Hg-Ga合金具有最小的法拉第反應電荷傳遞阻抗 Rct和最大的界面雙電層電容Cdlt,該合金電荷傳遞阻力最小,電極反應迅速,電化學活性最好。含 Mg5Ga2相的 Mg-Hg-Ga陽極具有最大的韋伯阻抗,此合金電極反應中反應物和產物的擴散系數最小,這與合金具有最大的電極表面腐蝕產物電阻 Rox有關,大量腐蝕產物的堆積阻礙電極反應的反應物和產物在電極表面的擴散,導致最大的濃差極化,因此合金的電化學活性降低。
第二相粒子的尺寸、形態和分布對鎂陽極的電流效率和表面溶解狀態也有比較明顯的影響,大小均勻、形態規則、數量適中的第二相粒子使鎂陽極表現出較好的電化學性能[52]。
若第二相粒子體積增大,則與鎂基體的接觸面積也相應增加,第二相作為陰極相導致其周圍鎂基體優先溶解,貢獻的陽極電流和時間持續增加,使得第二相粒子脫溶引起的相對電流效率損失減小,因而陽極電流效率增加,但尺寸較大的第二相粒子將加速陽極的局部溶解,使陽極表面溶解的均勻性降低。
若第二相粒子在晶界連續分布,則引起晶界的局部活化、優先溶解,使陽極保持較低的電流效率和較差的表面溶解均勻性。若第二相粒子在晶內彌散分布,則陽極的活化表現為第二相粒子隨鎂基體溶解—脫落和合金元素的溶解—再沉積,并且隨著第二相粒子之間間距增加,陽極活化的主導因素將由第二相粒子隨鎂基體的溶解—脫落逐漸轉變為合金元素的溶解—再沉積。兩者對陽極電流效率的貢獻大體相當,因而陽極電流效率維持基本不變,在這種情況下,只要基體中固溶的合金元素能保持足夠的活性,都將得到較均勻的表面溶解狀態。
若第二相粒子形狀規則,則第二相粒子促進鎂基體溶解的速度在各個方向上大體相等,因而優先溶解較為充分,第二相粒子脫落引起的陽極電流效率損失相對較小;若第二相粒子形狀很不規則,則因其與鎂基體的相互鑲嵌不容易脫落,陽極電流效率損失也小,因此第二相粒子由規則到不規則,陽極電流效率先下降而后升高。此外,第二相粒子越規則,其周圍鎂基體的溶解和自身脫溶越均勻,陽極表面的宏觀溶解必然也均勻。
馮艷等[8]通過研究第二相 Mg21Ga5Hg3和 Mg5Ga2顆粒的大小、形態和分布對Mg-Hg-Ga陽極電化學性能的影響,指出尺寸為10~20 nm,晶內彌散分布的規則球狀Mg21Ga5Hg3顆粒(見圖9(a))使Mg-Hg-Ga陽極具有最佳的電化學活性和耐腐蝕性能,而晶內析出的長條狀、塊狀Mg5Ga2第二相(見圖9(b))使Mg-Hg-Ga陽極電化學活性未得到明顯提高,而且耐腐蝕性能大大降低。

圖9 Mg-Hg-Ga陽極晶內析出相的形貌Fig.9 Morphologies of transgranular precipitates in Mg-Hg-Ga anodes
5 結語
鎂具有自然資源豐富、物理化學性能良好以及安全性能好的特點,是一種很有發展前景的高能量密度的電池陽極材料。近年來開發了適用于不同功率海水電池各種新型的鎂合金陽極,然而海水電池用鎂陽極的眾多研究領域仍缺乏深入的理論探討,如鎂電極在實際電池體系中的電化學過程動力學特征,鎂電極表面微觀電化學過程,電解液中離子的運動,眾多合金元素,如Zn、Bi、Pb、In的作用機理等。這些研究的不深入導致應用領域的有些問題難以從根本上得到解決,如鎂陽極效率的提高,激活狀態的改變以及析氫腐蝕的降低等。今后海水電池用鎂陽極的發展應在充分研究不同合金元素對鎂陽極的活化機理及其電化學動力學過程的基礎上,尋找陽極利用率高的鎂合金陽極材料,進一步減小析氫的腐蝕,解決活化與鈍化的矛盾,提高其比能量。
REFERENCES
[1] 宋玉蘇, 王樹宗. 海水電池研究及應用[J]. 魚雷技術, 2004,12(2): 4?8.SONG Yu-su, WANG Shu-zong. Research and application of seawater battery[J]. Torpedo Technology, 2004, 12(2): 4?8.
[2] ZHAO H Y, BIAN P, JU D Y. Electrochemical performance of magnesium alloy and its application on the sea water battery[J].Journal of Environmental Science Supplement, 2009, 21(1):S88?S91.
[3] 王樹宗. 魚雷動力電池技術發展水平概述[J]. 海軍工程學院學報, 1994(1): 95?105.WANG Shu-zong. Summary of development of torpedo propulsion technique[J]. Journal of Naval Academy of Engineering, 1994(1): 95?105.
[4] 蔡年生. 國外魚雷動力電池的發展及應用[J]. 魚雷技術, 2003,11(1): 12?16.CAI Nian-sheng. Development and application of batteries for overseas torpedo propulsion[J]. Torpedo Technology, 2003, 11(1):12?16.
[5] FONT S, DESCROIX J P, SARRE G. Advanced reserve batteries for torpedoes propulsion[C]//Cherry H. Proceedings of the 31st Power Sources Symposium. Penniton: Electrochemical Society,1984: 362?368.
[6] LI Q F, BJERRUM N J. Aluminum as anode for energy storage and conversion: a review[J]. Journal of Power Sources, 2002,110(1): 1?10.
[7] 馬正青, 左 列, 龐 旭, 曾蘇民. 鋁電池研究進展[J]. 船電技術, 2008, 28(5): 257?261.MA Zheng-qing, ZUO Lie, PANG Xu, ZENG Su-min. Advance in aluminum batteries[J]. Marine Electric and Electronic Technology, 2008, 28(5): 257?261.
[8] 馮 艷. Mg-Hg-Ga陽極材料合金設計及性能優化[D]. 長沙:中南大學, 2009.FENG Yan. Alloy design and properties optimization of Mg-Hg-Ga anode materials[D]. Changsha: Central South University, 2009.
[9] 彭成紅, 朱 敏. 鎂電池研究進展[J]. 電池, 2003, 33(2):121?123.PENG Cheng-hong, ZHU Min. Development of magnesium batteries[J]. Battery Bimonthly, 2003, 33(2): 121?123.
[10] YAMAMOTO O, KANBARA T, ITO S. Magnesium alloy battery: US 6265109[P]. 2001?07?24.
[11] HIROI M. Pressure effects on the performance and the e.m.f. of the Mg-AgCl seawater battery[J]. Journal of Applied Electrochemistry, 1980, 10(2): 203?211.
[12] 馬素卿, 陳亞昕. 水下推進用高能電池[J]. 船電技術, 1999,19(1): 13?23.MA Su-qing, CHEN Ya-xin. High-energy battery for underwater propulsion[J]. Marine Electric & Electronic Technology, 1999,19(1): 13?23.
[13] 奚碚華, 夏 天. 魚雷動力電池研究進展[J]. 魚雷技術, 2005,13(2): 7?12.XI Bei-hua, XIA Tian. Survey of power battery for torpedo propulsion[J]. Torpedo Technology, 2005, 13(2): 7?12.
[14] 姜憶初. 電動魚雷用動力電源及其發展方向[J]. 船電技術,2005, 25(5): 46?48.JIANG Yi-chu. Electric power sources used in electric torpedo and its development trends[J]. Marine Electric and Electronic Technology, 2005, 25(5): 46?48.
[15] 王樹宗. A244/s魚雷動力電池材料與制造工藝分析[J]. 魚雷技術, 1994, 2(1) : 46?53.WANG Shu-zong. Analysis for manufacture process of A244/s torpedo power battery materials[J]. Torpedo Technology, 1994,2(1): 46?53.
[16] FENG Y, WANG R C, YU K, PENG C Q, ZHANG J P, ZHANG C. Activation of Mg-Hg anodes by Ga in NaCl solution[J].Journal of Alloys and compounds, 2009, 473(1/2): 215?219.
[17] 鄧姝皓, 易丹青, 趙麗紅, 周玲伶, 王 斌, 冀成年, 蘭 博.一種新型海水電池用鎂負極材料的研究[J]. 電源技術, 2007,31(5): 402?405.DENG Su-hao, YI Dan-qing, ZHAO Li-hong, ZHOU Ling-ling,WANG Bin, JI Cheng-nian, LAN Bo. Study on Mg alloy anode material for seawater battery[J]. Battery Technology, 2007, 31(5):402?405.
[18] CAO F H, LEN V H, ZHANG Z, ZHANG J Q. Corrosion behavior of magnesium and its alloy in NaCl solution[J].Russian Journal of Electrochemistry, 2007, 43(7): 837?843.
[19] UHLENHAUT D I, FURRER A, UGGOWITZER P J,LOFFLER J F. Corrosion properties of glassy Mg70Al15Ga15 in 0.1 M NaCl solution[J]. Intermetallics, 2009, 17(10): 811?817.
[20] WANG Lei, ZHANG Bo-ping, TADASHI S. Corrosion behavior of AZ91 magnesium alloy in dilute NaCl solutions[J]. Materials and Design, 2010, 31(2): 857?863.
[21] 楊 武. 金屬的局部腐蝕?點腐蝕·縫隙腐蝕·晶間腐蝕·成分選擇性腐蝕[M]. 北京: 化工出版社, 1995.YANG Wu. Local corrosion of metal pitting corrosion· crevice corrosion· intergranular corrosion· composition selective corrosion[M]. Beijing: Chemical Industry Press, 1995.
[22] 侯軍才. 高電位鎂陽極的制備及性能研究[D]. 鄭州: 鄭州大學, 2006.HOU Jun-cai. Preparation and performance research of high-potential magnesium anode[D]. Zhenzhou: University of Zhenzhou, 2006.
[23] 余 琨, 黎文獻, 李松瑞. 變形鎂合金材料研究進展[J]. 輕合金加工技術, 2001, 29(7): 6?10.YU Kun, LI Wen-xian, LI Song-rui. The research and developments of wrought magnesium alloy[J]. Light Alloy Fabrication Technology, 2001, 29(7): 6?10.
[24] BENDER S, GOELLNER J, ATRENS A. Corrosion of AZ91 in 1 M NaCl and the mechanism of magnesium corrosion[J].Advanced Engineering Materials, 2008, 10(6): 583?587.
[25] PARDO A, MERINO M C, COY A E, ARRABAL R, VIEJO F,MATYKINA F. Corrosion behaviour of magnesium/aluminium alloys in 3.5 wt.% NaCl[J]. Corrosion Science, 2008, 50(3):823?834.
[26] HANAWALT J D, NELSON C E, PELOUBET J A. Corrosion studies of magnesium and its alloys[J]. Transaction of American Institute of Mining, Metallurgical, and Petroleum Engineers,1942, 147: 273?299.
[27] FENG Y, WANG R C, YU K, LI W X. Influence of heat treatment on electrochemical behavior of Mg anode materials[J].Journal of Central South University of Technology, 2007, 2(14):12?15.
[28] SONG G L, ATRENS A. Corrosion mechanisms of magnesium alloys[J]. Advanced Engineering Materials, 1999, 1(1): 11?33.
[29] MAKAR G L, KRUGER K. Corrosion of magnesium[J].International Materials Reviews, 1993, 38(3): 138?153.
[30] MAKAR G L, KRUGER K. Corrosion studies of rapidly solidified magnesium alloys[J]. Journal of Electrochemical Society, 1990, 137(2): 414?421.
[31] UHLIG H H, REVIE R W. Corrosion and corrosion control[M].3rd ed. New York: WILEY J, 1985.
[32] SONG G, ATRENS A, STJOHN D, NAIRN J, LI Y. The electrochemical corrosion of pure magnesium in 1 M NaCl[J].Corrosion Science, 1997, 39(5): 855?875.
[33] LUNDER O, NISANCIOGLU K, HANSEN R S. Corrosion of die cast magnesium-aluminum alloys, SAE Technical Paper Series #930755[R]. Detroit, 1993.
[34] HOEY G R, COHEN M. Corrosion of anodically and cathodically polarized magnesium in aqueous media[J]. Journal of Electrochemical Society, 1958, 105(5): 245?250.
[35] SONG G L, ATRENS A, WU X, BO Z, ZHANG B. Corrosion behaviour of AZ21, AZ501 and AZ91 in sodium chloride[J].Corrosion Science, 1998, 40(10): 1769?1791.
[36] SONG G L, ATRENS A, DARGUSCH M. Influence of microstructure on the corrosion of diecast AZ91D[J]. Corrosion Science, 1999, 41(2): 249?273.
[37] SONG G L, ATRENS A. Recent progress in corrosion and protection of magnesium alloys-An over of cast’s research work[J]. Advanced Engineering Materials, 2005, 7(7): 563?586.[38] SONG G L, ATRENS A. Understanding magnesium corrosion-a framework for improved alloy performance[J]. Advanced Engineering Materials, 2003, 5(12): 837?858.
[39] SONG G L. Investigation on corrosion of magnesium and its alloys[J]. The Journal of Corrosion Science and Engineering,2007, 6: C104.
[40] 宋光鈴. 鎂合金腐蝕與防護[M]. 北京: 化學工業出版社,2006.SONG Guang-ling. Corrosion and protection of magnesium[M].Beijing: Chemical Industry Press, 2006.
[41] LIU L J, SCHLESINGER M. Corrosion of magnesium and its alloys[J]. Corrosion Science, 2009, 51(8): 1733?1737.
[42] 袁華堂, 吳 峰, 武緒麗, 李 強. 可充鎂電池的研究和發展趨勢[J]. 電池, 2002, 32(Z1): 14?17.YUAN Hua-tang, WU Feng, WU Xu-li, LI Qiang. The study and development of rechargeable magnesium battery[J]. Battery Nimonthly, 2002, 32(Z1): 14?17.
[43] FENG Yan, WANG Ri-chu, YU Kun, PENG Chao-qun, LI Wen-xian. Influence of Ga and Hg on microstructure and electrochemical corrosion behavior of Mg alloy anode materials[J]. Transactions of Nonferrous Metals Society of China,2007, 17(6): 1363?1366.
[44] FENG Y, WANG R C, YU K, PENG C Q, LI W X. Influence of Ga content on electrochemical behavior of Mg-5at% Hg anode materials[J]. Materials Transactions, 2008, 49(5): 1077?1080.
[45] SAIDMAN S B, BESSONE J B. Cathodic Polarization characteristics and activation of aluminum in chlorid solution containing indium and zinc ions[J]. Journal of Applied Electrochemistry, 1997, 27(6): 731?737.
[46] SHAYEB H A, WAHAB F M A, ABEDIN S Z. Electrochemical behaviour of Al, Al-Sn, Al-Zn, Al-Sn-Zn alloys in chloride solution containing stannousions[J]. Corrosion Science, 2001,43(4): 655?669.
[47] 候德龍, 宋月清, 王 譯, 李德富, 何德山. 高電位鎂合金(Mg-Mn)陽極熔體凈化技術的研究[J]. 稀有金屬, 2006, 30(1):30?33.HOU De-long, SONG Yue-qing, WANG Yi, LI De-fu, HE De-shan. Purification technology of Mg-Mn alloy sacrificial anodes[J]. Rare Metal, 2006, 30(1): 30?33.
[48] JUNG G K, JIN H J, SE J K. Development of high-driving potential and high-efficiency Mg-based sacrificial anodes for cathodic protection[J]. Journal of Materials Science Letters,2000, 19(6): 477?479.
[49] JUNG G K, KIM Y W. Advanced Mg-Mn-Ca sacrificial anode materials for cathodic protection[J]. Materials and Corrosion,2001, 52(2): 137?139.
[50] BELDJOUDI T, FIAUD C, ROBBIOLA L. Influence of homogenization and artificial aging heat treatments on corrosion behavior of Mg-Al alloys[J]. Corrosion Science, 1993, 49(9):738?745.
[51] KING J F, UNSWORTH W. Magnesium in seawater batteries[J].Light Metal Age, 1978, 36(7/8): 22?24.
[52] ANIK M, AVCI P, TANVERDI A, CELIKYUREK I, BAKSAN B, GURLER R. Effect of the eutectic phase mixture on the anodic behavior of alloy AZ91[J]. Materials and Design, 2006,27(5): 347?355.
Researches and applications of magnesium anode materials in seawater battery
FENG Yan, WANG Ri-chu, PENG Chao-qun
(School of Materials Science and Engineering, Central South University, Changsha 410083, China)
The magnesium anodes have great development in seawater battery due to their excellent properties, such as good electro negativity, high specific capacity and low density. The researches and applications of magnesium anodes in seawater battery in recent years were reviewed. The influences of activation mechanism, corrosion behavior, alloying elements and second phases on the electrochemical and corrosion properties of magnesium anodes were discussed. It is indicated that the development of magnesium anodes in seawater battery is to prepare novel anode materials with less self-corrosion, higher anode utilization ratio and larger specific energy based on studying activation mechanism of alloying elements and second phases.
seawater battery; magnesium anode; activation; corrosion; alloying element; second phase
TG174.451
A
1004-0609(2011)02-0259-10
國防科工委民口配套研制項目(MKPT-02-181)
2009-12-01;
2010-03-16
王日初,教授,博士;電話:0731-88836638;E-mail:wrc910103@163.com
(編輯 李艷紅)
