環烷酸模型化合物酯化反應體系的熱力學分析
2011-10-13 03:24:14任正時朱建華
天津化工 2011年2期
任正時,朱建華
(中國石油大學化學科學與工程學院,北京102249)
環烷酸模型化合物酯化反應體系的熱力學分析
任正時,朱建華
(中國石油大學化學科學與工程學院,北京102249)
以選取的環烷酸模型化合物為基礎,利用熱力學方法分析其酯化反應的可能性。由于環烷酸物質結構的特殊性,它們的熱力學數據通常無法直接獲取,采用了基團貢獻法估算酯化反應體系中環烷酸及環烷酸酯在298.15K標準態下的生成焓和標準熵,同時確定了這兩種物質等壓熱容隨溫度的變化關系,進而得到了不同溫度條件下酯化反應的吉布斯自由能變及平衡常數。計算結果表明,酯化反應的平衡常數很大(107),從而確定了該反應體系的可能性。
環烷酸;酯化脫酸;熱力學分析;基團貢獻法
環烷酸是原油中存在的石油酸的主要成分,其基本結構為含有五元環或六元環的單環或多環一元羧酸。由于在原油加工過程中環烷酸腐蝕設備,導致設備使用周期減短[1],如何減少其對設備的腐蝕,并使之轉化為有用的成分,是一個亟待解決的問題。本文選取帶有雙環的-(3,4)-環戊烷基環己烷乙酸作為環烷酸的模型化合物,同時為了使生成的環烷酸酯成為餾分油的有益成分,選取乙二醇作為進行酯化反應的醇類物質。在常壓及一定的溫度條件下,-(3,4)-環戊烷基環己烷乙酸與過量乙二醇在催化劑作用下的酯化反應。
1 熱力學數據的估算
1.1 298.15 K理想狀態條件下生成焓與規定熵值的估算
利用Benson基團貢獻法[2~4],估算反應體系中環烷酸(酸C11H18O2)和環烷酸酯(酯C13H22O3)在298.15K標準狀態下為理想氣體時的生成焓和標準熵值的計算公式如下所示:
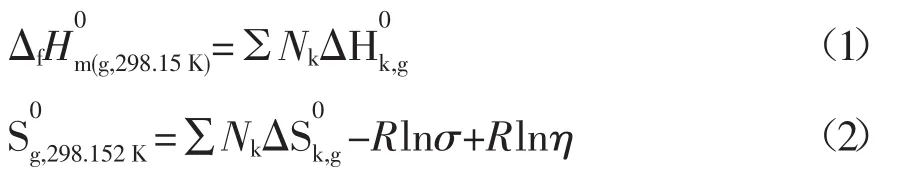
式中:Nk為k型基團的數目;σ為分子的對稱數;η為光學異構體數。
反應組分中各基團的貢獻值如表1所示:

表1 Benson基團貢獻值
將表1中的數據代入式(1)和(2)中,便可計算出各物質在298.15 K標準狀態下呈理想氣體狀態時的摩爾生成焓與摩爾標準熵的估算值,計算結果如表2所示:
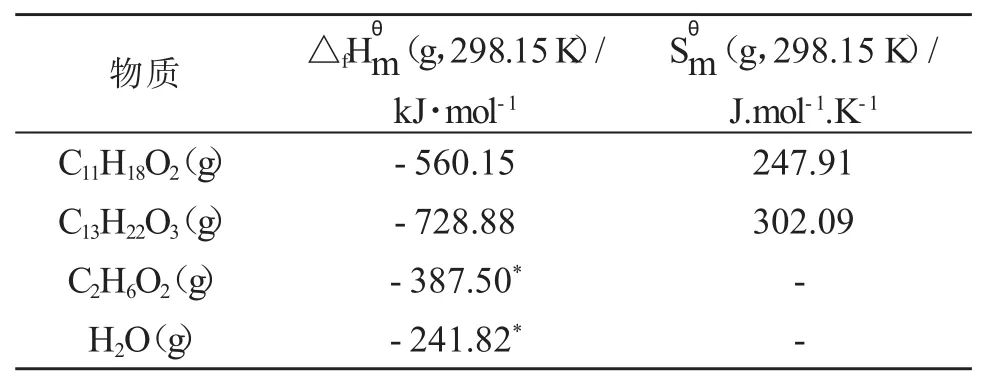
表2 標準狀態下各組分的標準摩爾生成焓和標準熵物質
1.2 298.15K條件下各組分液態生成焓的估算
由于反應體系中的各組分均以液態形式參與反應,所以需對各組分在298.15 K呈液態狀態下的相關熱力學數據進行估算,298.15 K時各組分呈液態時的焓變按式(3)計算:


式中:ni表示i型基團的數目;εi表示i型基團對蒸發熱的貢獻值,單位為J·mol-1。
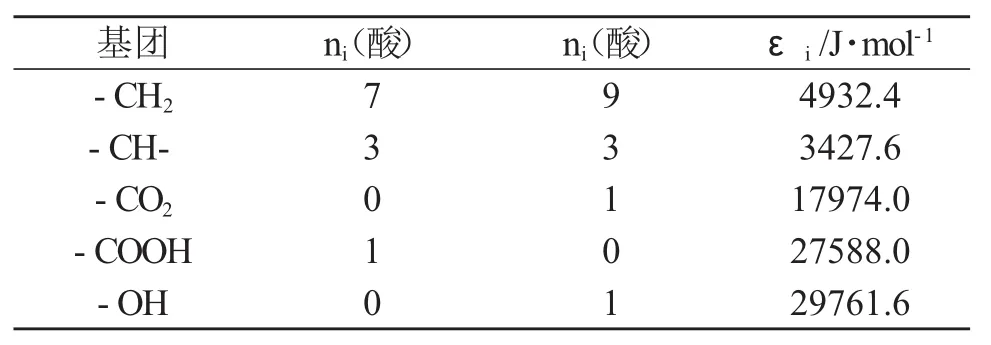
表3 Fedors基團貢獻值
298.15K各組分為理想液體時的標準摩爾生成焓計算結果為:C11H18O2(l)-635.026 kJ·mol-1、C13H22O3(l)-833.768 kJ·mol-1、C2H6O2(l)-455.340*kJ·mol-1、H2O(l)-285.830*kJ·mol-1。帶*的為文獻[6]數據。
1.3 298.15 K條件下液態組分規定熵值的估算
在298.15 K標準狀態下,酯化反應中各組分呈液態時的規定熵值可由式(5)計算:
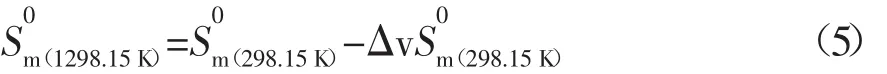
式中:為液態組分在298.15 K標準狀態下的摩爾蒸發熵,可按經驗關聯式(6)進行計算[2,3]:

式中:A和B均為經驗常數,二者的數值應根據化合物的類型進行選取,估算酸或醇時,A、B分別取390.203 J·mol-1·K-1和-873.3274 J·mol-1·K-1;估算酯時,A、B分別取125.4 J·mol-1·K-1和-222.376 J·mol-1·K-1;Tb為物質在常壓下的沸點。據相關文獻[5]可知乙二醇的沸點為505.15 K,由Joback基團貢獻法[2,3]可估算出-(3,4)-環戊烷基環己烷乙酸及-(3,4)-環戊烷基環己烷乙酸2-羥基乙酯的沸點分別為618.21 K和668.86K。
結合式(5)和(6)可以計算出在298.15 K標準狀態下各組分的摩爾蒸發熵和298.15 K條件下呈液態時的規定熵值,結果見表4。

表4 298.15K各組分為理想液體時的標準摩爾規定熵
1.4 液態有機組分熱容的估算
當溫度低于沸點時,不同溫度條件下各組分液態熱容的估算可采用Ruzicka-Domalski基團貢獻法[2~6],計算公式如式(7)所示:
我國柳屬植物資源豐富,但目前對其研究多屬于基礎性藥理研究,臨床應用研究較少,藥用資源的開發利用還比較薄弱。今后應進一步在中醫藥理論的指導下,對柳屬植物進行系統的藥效活性篩選和化學成分分離,從中尋找活性強、療效高、毒副作用小的天然產物進行藥品的研發;利用構效關系對天然化合物進行結構修飾,開發更適于臨床的高效低毒的藥物;結合分子生物學對藥物的作用靶點進行預測,深入研究其作用機制;對活性部位及活性化合物進行臨床研究,為柳屬植物臨床用藥的安全性提供可靠的科學依據,進而促進柳屬植物的合理開發與應用。
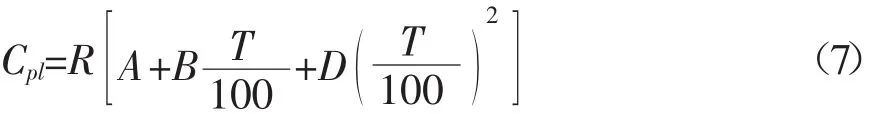
各組分的Ruzicka-Domalski基團貢獻值如表5所示:
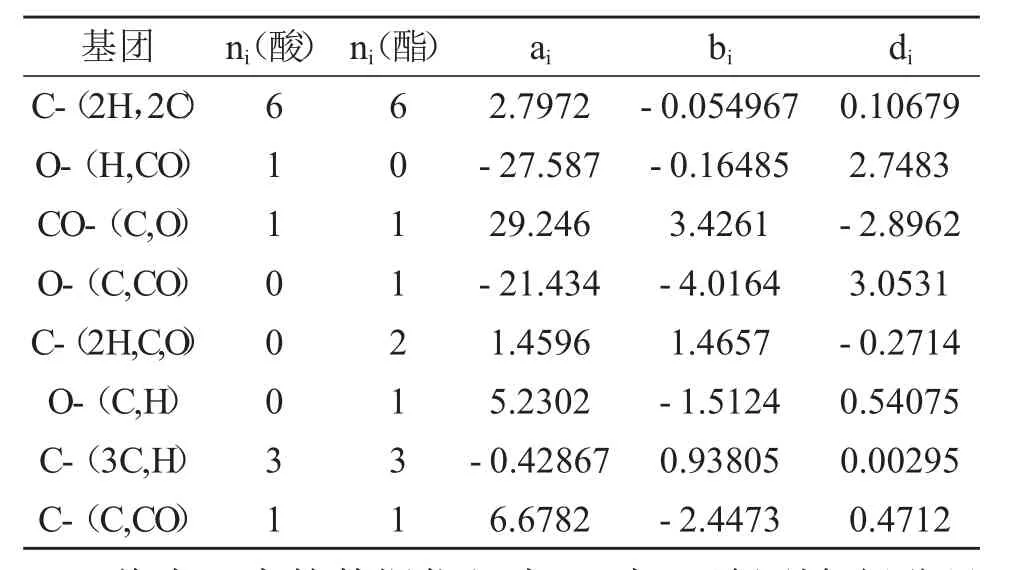
表5 Ruzicka-Domalski基團貢獻值
將表5中的數據代入式(7)中,可得到各組分呈液態時的熱容表達式(J·mol-1·K-1),結果是:C11H18O2(l)為198.159+0.274T+8.089×10-4T2、C13H22O3(l)為317.069+0.072T+1.061×10-3T2、C2H6O2(l)為111.238-7.765×10-3T+4.479×10-4T2、H2O(l)*為75.6(溫度在298.15~373.15 K范圍內的平均值)、H2O(g)*為29.16+14.19×10-3T-0.2022×10-5T2(溫度高于373.15 K時)。帶*的為文獻[7]數據。
2 熱力學分析
2.1.298.15-373.15 K范圍內時
由各組分的熱容與溫度關系式,可求得在298.15-373.15 K溫度范圍內該反應的熱容(單位:J·mol-1·K-1)表達式如下:
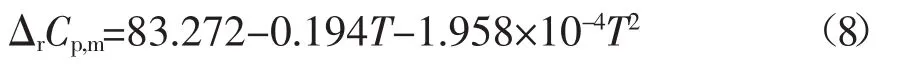
將298.15 K時反應的焓變值代入Kirchhoff公式[7]:[?(ΔrG/T)/?T]p=-ΔrH/T2,可得到在此溫度區間內反應焓變(單位:J·mol-1)隨溫度變化的關系式如下:
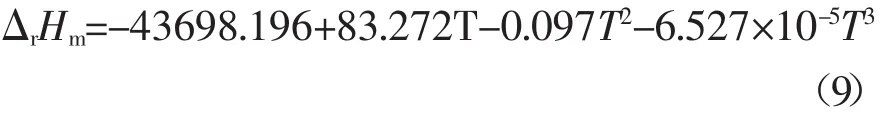
將298.15K時反應的吉布斯自由能的變化值代入Gibbs-Helmholtz方程[7]:[?(ΔrG/T)/?T]p=-ΔrH/T2,可計算得到在此溫度區間內反應的吉布斯自由能變(單位:J·mol-1)隨溫度變化的關系式如下:

2.2 在373.15-450 K范圍內時
在373.15K、常壓條件下,水的摩爾蒸發焓為40.637 kJ·mol-1,該可逆過程的摩爾蒸發熵為108.903 J·mol-1·K-1,通過恒壓變溫過程和可逆過程設計,并結合相關的熱力學公式,可計算得到在373.15 K、標準狀態下該反應的焓變、熵變和吉布斯自由能變分別為:11.098 kJ·mol-1、162.655 J·mol-1·K-1、-49.60 kJ·mol-1。
根據各組分熱容與溫度的關系,可求得在373.15-450K溫度范圍內反應的熱容表達式如下:
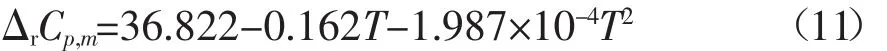
同理可得到373.15-450 K溫度范圍內反應的焓變和自由能變與溫度的關系式分別如下所示:

通過式(9)、(10)、(12)和(13)可計算得到不同溫度條件下,反應的焓變、吉布斯自由能變和平衡常數值,其中反應平衡常數按求得,根據計算結果繪制出圖1、圖2、圖3。
3 結果與討論
由圖1可知,反應過程中生成水的相態改變對反應熱的影響非常顯著。以常壓條件下水的沸點為界,在298.15-373.15 K溫度區間內該反應的焓變均小于零,表明反應為放熱反應,從熱力學上講此時低溫對反應有利;而在溫度大于373.15K時,反應的焓變均大于零,此時該反應為吸熱反應,升高溫度將有利于反應向生成酯的方向移動。
由圖2可知,在溫度區間內該反應的Gibbs自由能變均小于零,說明在此溫度區間內,該反應自發進行,從而判知該酯化反應在指定的反應條件下是可能發生的。而在實際反應過程中,可使用高活性的催化劑加速該酯化反應,縮短酯化反應達到平衡所需的時間。
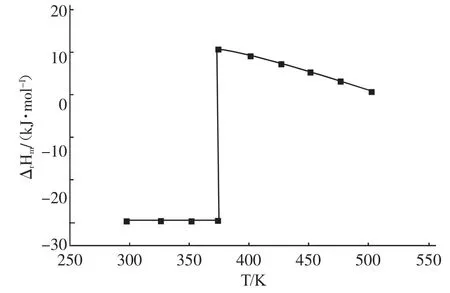
圖1 反應焓變與溫度的關系

圖2 反應Gibbs自由能變與溫度的關系
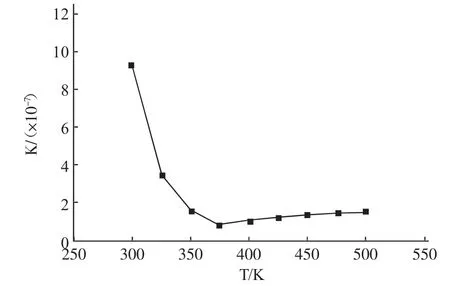
圖3 反應平衡常數與溫度的關系
由圖3可知,在298.15~373.15 K溫度區間內,反應平衡常數隨溫度升高而急劇減小;當溫度高于373.15 K時,反應平衡常數隨溫度的升高而增大,這是由于在兩個溫度范圍內反應熱效應的不同引起的。在整個考察溫度范圍內,反應平衡常數值均非常大,因此在此條件下當反應達到平衡時,反應幾乎能夠進行到底,這表明該反應在熱力學上是完全可能的。另外,由于該反應是一個典型的可逆反應,在反應過程中若能將生成的水及時移出反應體系,將會使平衡向生成酯的方向移動,并縮短反應達到平衡所需的時間。因此,在合成環烷酸酯的過程中,需采取行之有效的方法將反應生成的水及時從體系中分離出去,推動反應向生成環烷酸酯的方向進行以實現酯化反應過程的強化。
4 關于合成環烷酸酯化反應的副反應
由于該反應體系中的-(3,4)-環戊烷基環己烷乙酸在反應溫度內是穩定的,副反應主要為乙二醇的分子內和分子間脫水以及環烷酸酯與乙二醇生成環烷酸二酯的反應,它們均是互相競爭的反應。副反應的發生可能是由于乙二醇過量和溫度引起的,因此在反應過程中需控制加入乙二醇的量及適宜的反應溫度。此外選擇合適的催化劑對抑制各種副反應的發生也非常重要。
5 小結
通過對環烷酸與乙二醇酯化反應體系的熱力學分析,可以得到以下結論:
(1)采用Benson,Fedors,Rùzicka-Domalski和Joback基團貢獻法對-(3,4)-環戊烷基環己烷乙酸、乙二醇和-(3,4)-環戊烷基環己烷乙酸2-羥基乙酯在298.15 K標準狀態下的熱力學數據進行了估算,為該酯化反應的熱力學分析提供了基礎數據。
(2)確定了不同溫度區間內反應的熱容、焓變、Gibbs自由能,及反應平衡常數與溫度的關系式,從而為-(3,4)-環戊烷基環己烷乙酸與乙二醇酯化反應體系的熱力學分析提供了依據。
(3)熱力學分析結果表明,該酯化反應在所分析的溫度區間可自發進行,且具有較大的平衡常數,從而驗證了環烷酸模型化合物與乙二醇發生酯化反應的可能性。此外,對該反應體系中可能發生的副反應也進行了簡要分析。
由于大部分環烷酸均為同系物,對于其它環烷酸與二元醇的酯化反應,可依據本文建立的方法進行熱力學分析,從而為高酸原油酯化脫酸技術的開發,并將高酸原油中的環烷酸轉化為有益組分—環烷酸酯提供了理論指導。
[1]呂振波,田松柏,翟玉春,等.原油中環烷酸腐蝕預測方法綜述[J].石油化工腐蝕與防護,2004,21(3):1-4.
[2]劉光啟,馬連湘,劉杰.化學化工物性數據手冊[M].北京:化學工業出版社,2002.
[3]馬沛生.化工數據[M].北京:中國石化出版社,2003.
[4]董新法,方利國,陳礪.物性估算原理及計算機計算[M].北京:化學工業出版社,2006.
[5]PolingBE,Prausnitz J M,John.P.O'Connell.The Properties of Gases and Liquids,5th ed.[M].New York:McGraw-Hill Companies,Inc.,2001.
[6]李夢龍.化學數據速查手冊[M].北京:化學工業出版社,2003.
[7]宋世謨,莊公惠,王正烈.物理化學(第三版)[M].北京:高等教育出版社,1992.
Thermodynamics analysis for the esterification of model naphthenic acid compounds
REN Zheng-shi,ZHU Jian-hua
(Faculty of Chemical Science and Engineering,China University of Petroleum,Beijing102249,China)
In this paper,the possibility of the esterification of model naphthenic acid compounds in high acid oil would be verified by thermodynamic analysis method.Due to the particularity of these organic compounds,their thermodynamic date could not be found in literatures.Therefore,the methods of group contribution are applied to estim ate the enthalpy and entropy of naphthenic acid,ethylene glycol and naphthenic acid esters under the standard state.Meanwhile,the functional correlations between the heat capacity of these organic compounds and temperature are established.Furthermore,the Gibbs free energy change and equilibrium constant of the esterification reaction are calculated under the different temperature.The result shows that equilibrium constant of the reaction are very large(>107) which prove the esterification reaction of naphthenic acid and ethylene glycol is possible in view of thermodynamics.
naphthenic acid;esterification;thermodynamic analysis;group contribution method.
10.3969/j.issn.1008-1267.2011.02.009
O624.5
A
1008-1267(2011)02-0022-05
2010-11-02
任正時(1984-),男,湖北荊州人,碩士研究生,主要從事原油深度預處理研究。朱建華(1963-),男,博士生導師,中國石油大學(北京)教授。
