冠心生脈口服液質量標準研究
2011-09-17 06:40:34李海燕
中國藥業 2011年1期
李海燕,康 建
(1.河南省食品藥品檢驗所,河南 鄭州 450003; 2.鄭州大學第一附屬醫院,河南 鄭州 450052)
冠心生脈口服液被收載于《衛生部藥品標準新藥轉正標準(第十冊)》,具有益氣生津、活血通脈的功效,用于治療心氣不足、心陰虛引起的心悸氣短、胸悶作痛、自汗乏力、脈微結代。全方由人參、麥冬、五味子(醋炙)、丹參、赤芍、郁金、三七等藥材組成。原標準中的質量控制方法較簡單且操作煩瑣,不能有效控制藥品質量。筆者根據處方所含藥味的化學成分及劑型特點,研究制訂了赤芍、麥冬、人參及三七、五味子的薄層色譜鑒別及赤芍中芍藥苷的含量測定[1]方法。現報道如下。
1 儀器與試藥
Waters 2690型高效液相色譜儀;Waters 996型二極管陣列檢測器;硅膠G預制薄層板(青島海洋化工廠,批號為030507);超聲波清洗機(功率250 W,頻率40 kHz)。麥冬、人參、三七對照藥材(批號分別為 121013-200607,120917-200609,120941-200506)及芍藥苷對照品(批號為 110736-200731,供含量測定用,以97.9%計算)、五味子醇甲對照品(批號110857-200608)均由中國藥品生物制品檢定所提供;冠心生脈口服液(批號分別為081001,081002,081003,河南省宛西制藥股份有限公司);甲醇、乙腈為色譜純,水為樂百氏純凈水,其他試劑均為分析純。
2 方法與結果
2.1 薄層色譜鑒別
赤芍:取本品10 mL,置分液漏斗中,用正丁醇10 mL振搖提取,將正丁醇液蒸干,殘渣加甲醇1 mL使溶解,作為供試品溶液;按冠心生脈口服液制備工藝制備不含赤芍的陰性對照品溶液;另取芍藥苷對照品,加甲醇制成每1 mL含1 mg的溶液,作為對照品溶液。照薄層色譜法試驗,吸取上述3種溶液各10 μL,分別點于同一硅膠G薄層板上,以三氯甲烷-乙酸乙酯-甲醇-甲酸(40 ∶5 ∶10 ∶0.2)為展開劑,展開,取出,晾干,噴以 5% 香草醛硫酸溶液,加熱至斑點顯色清晰。結果供試品溶液色譜中,在與對照品溶液色譜相應位置上顯相同顏色的斑點,陰性對照品溶液無干擾(圖 1 A)。
麥冬:取本品10 mL,加鹽酸溶液(18→100)1 mL,置水浴上濃縮至約5 mL,用三氯甲烷10 mL振搖提取,將三氯甲烷液濃縮至2 mL,作為供試品溶液;按冠心生脈口服液制備工藝制備不含麥冬的陰性對照品溶液;另取麥冬對照藥材1 g,加水30 mL,加熱回流30 min,濾過,濾液濃縮至10 mL,同法制成對照藥材溶液。照薄層色譜法試驗,吸取上述3種溶液各10 μL,分別點于同一硅膠G薄層板上,以三氯甲烷-丙酮(4∶1)為展開劑,展開,取出,晾干,噴以10%硫酸乙醇溶液,加熱至斑點顯色清晰。結果供試品溶液色譜中,在與對照藥材溶液色譜相應位置上顯相同顏色的斑點,陰性對照品溶液無干擾(圖1 B)。
人參及三七:取本品30 mL,用乙酸乙酯30 mL振搖提取,水液用水飽和的正丁醇提取振搖2次,每次30 mL,合并正丁醇液,用氨試液洗滌2次,每次30 mL,正丁醇液用正丁醇飽和的水30 mL洗滌,正丁醇液蒸干,殘渣加甲醇1 mL使溶解,作為供試品溶液;按冠心生脈口服液制備工藝制備不含人參和三七的陰性對照品溶液;另取人參、三七對照藥材各1 g,分別加甲醇25 mL,加熱回流1 h,放冷,濾過,濾液蒸干,殘渣加水20 mL使溶解,分別同法制成對照藥材溶液。照薄層色譜法試驗,吸取供試品溶液5 μL、人參對照藥材溶液及三七對照藥材溶液各1 μL,分別點于同一硅膠G薄層板上,以10℃下放置的三氯甲烷-甲醇-水(13∶7∶2)的下層溶液為展開劑,在2~10℃展開,取出,晾干,噴以10%硫酸乙醇溶液,在105℃加熱至斑點顯色清晰。結果供試品溶液色譜中,在與對照藥材溶液色譜相應位置上分別顯相同顏色的斑點,陰性對照品溶液無干擾(圖1 C)。
五味子:取本品50 mL,用三氯甲烷振搖提取2次,每次30 mL,將三氯甲烷液蒸干,殘渣加三氯甲烷1 mL使溶解,作為供試品溶液;按冠心生脈口服液制備工藝制備不含五味子的陰性對照品溶液;另取五味子醇甲對照品,加甲醇制成每1 mL含1 mg的溶液,作為對照品溶液。照薄層色譜法試驗,吸取供試品溶液10 μL,對照品溶液1 μL,分別點于同一硅膠GF254薄層板上,以石油醚(30~60℃)-甲酸乙酯-甲酸(10∶5∶1)的上層溶液為展開劑,展開,取出,晾干,置紫外光燈(254 nm)下檢視。結果供試品溶液色譜中,在與對照品溶液色譜相應位置上顯相同顏色的熒光斑點,陰性對照品溶液無干擾(圖1 D)。
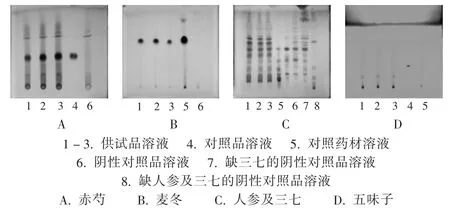
圖1 薄層色譜圖
2.2 含量測定
2.2.1 色譜條件與系統適用性試驗
色譜柱:Agilent Eclipse XDBC18柱 (250 mm ×4.6 mm,5 μm);柱溫:35℃;檢測波長:230 nm;流動相:乙腈-水(14∶86)[2];流速:1 mL/min;進樣量:10 μL。理論板數按芍藥苷峰計算應不低于5 000。在此色譜條件下的色譜圖見圖2。
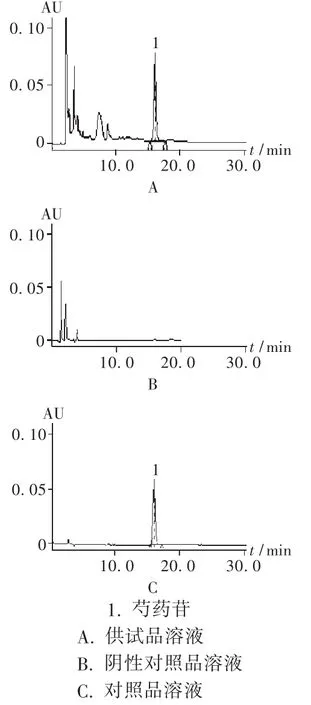
圖2 高效液相色譜圖
2.2.2 溶液制備
精密稱取芍藥苷對照品適量,精密稱定,加甲醇制成每1 mL含0.1 mg的對照品溶液;精密量取本品 10 mL,置 100 mL量瓶中,加30%甲醇稀釋至刻度,搖勻,作為供試品溶液;按冠心生脈口服液制備工藝和供試品溶液制備方法制備不含赤芍的陰性對照品溶液。
2.2.3 方法學考察
線性關系考察:精密吸取芍藥苷對照品溶液(406.5 μg/mL)1,3,5,7,10,13 μL,按上述色譜條件測定。以進樣量(X)為橫坐標、峰面積積分值(Y)為縱坐標繪制標準曲線,得回歸方程 Y=1 445 284X-47 609,r=0.999 3(n=6)。結果表明,芍藥苷進樣量在 0.406 5 ~5.284 5 μg范圍內與峰面積線性關系良好。
精密度試驗:精密吸取同一供試品溶液10 μL,重復進樣6次,測定峰面積。結果的 RSD為0.09%(n=6)。
重復性試驗:對同一批(批號為081001)樣品5份進行測定。結果的 RSD為0.33%(n=5),表明方法重復性良好。
穩定性試驗:取同一供試品溶液,按上述色譜條件分別于0,2,4,6,8,10 h時測定芍藥苷峰峰面積積分值。結果的 RSD為0.34%(n=6),表明供試品溶液在10 h內基本穩定。
加樣回收試驗:取已知含量的同一批(批號為081001)樣品6份各5 mL,每份加對照品貯備液適量,按供試品溶液制備方法制備加樣供試品溶液,進樣測定,計算加樣回收率。結果見表1。
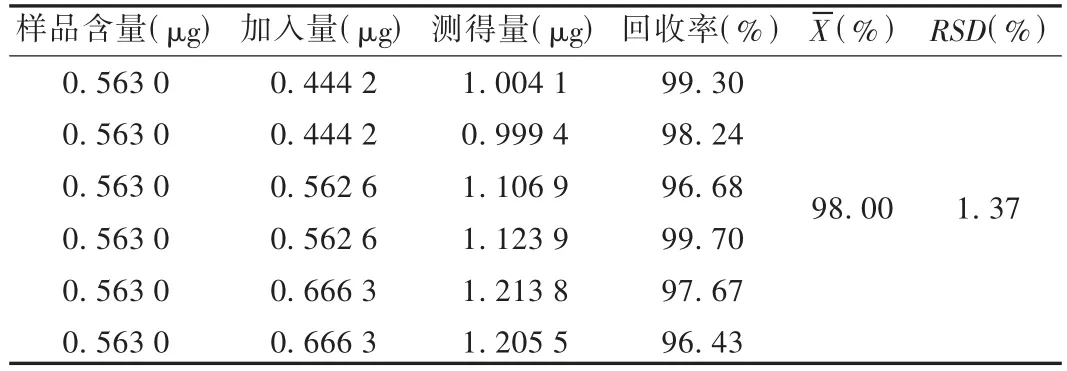
表1 芍藥苷加樣回收試驗結果(n=6)
2.2.4 樣品含量測定
分別取批號為 081001,081002,081003的 3批樣品,按“2.2.2”項下方法制成供試品溶液,進樣測定,按外標法計算含量。結果 3 批樣品中芍藥苷含量分別為 1.126,1.135,1.126 g/L。
3 討論
本品既含人參又含三七,用普通硅膠G薄層板無法對二者進行鑒別。筆者曾嘗試鑒別三七,以三七皂苷R1和三七對照藥材為對照,使用高效硅膠G薄層板(青島海洋化工廠),用10℃以下放置的三氯甲烷-甲醇-水(13∶7∶2)的下層溶液為展開劑,在2~10℃展開,還做了二次展開試驗,但分離效果和重現性不佳。因此,以人參、三七對照藥材同時作為對照,用普通硅膠G薄層板做鑒別試驗;試驗中曾以五味子對照藥材為對照,結果供試品溶液色譜中在與缺五味子的陰性對照的相應位置上有相同顏色的主斑點出現,陰性對照有干擾。
含量測定選用溶液直接稀釋的方法,操作簡易。比較了30%甲醇溶液、50%甲醇溶液、70%甲醇溶液及甲醇的提取效果。用甲醇作溶劑時,樣品液渾濁,用30%甲醇溶液、50%甲醇溶液及70%甲醇溶液提取時,樣品液澄清,因此選用30%甲醇溶液為提取溶劑。
[1]王亞林.HPLC測定宮炎康顆粒中芍藥苷的含量[J].陜西中醫學院學報,2006,29(4):41.
[2]國家藥典委員會.中華人民共和國藥典(一部)[M].北京:化學工業出版社,2005:405.
