微波輻射對漿態床合成甲醇CuO/ZnO/Al2O3催化劑前驅體晶相轉變的影響
2011-09-15 11:44:36忠鄭華艷閆少偉
無機化學學報 2011年3期
關鍵詞:催化劑
范 輝 李 忠鄭華艷 劉 巖 閆少偉
(太原理工大學煤化工研究所煤科學與技術教育部和山西省重點實驗室,太原 030024)
微波輻射對漿態床合成甲醇CuO/ZnO/Al2O3催化劑前驅體晶相轉變的影響
范 輝 李 忠*鄭華艷 劉 巖 閆少偉
(太原理工大學煤化工研究所煤科學與技術教育部和山西省重點實驗室,太原 030024)
在微波輻射條件下,對CuO/ZnO/Al2O3催化劑的沉淀母液進行老化,通過XRD、TG、H2-TPR,FTIR、HR-TEM和XPS對前驅體及催化劑微觀結構的進行表征,探討了CuO/ZnO/Al2O3催化劑前驅體晶相轉變過程中微波輻射的作用。結果表明,微波輻射有利于Cu2+取代Zn5(CO3)2(OH)6中Zn2+的同晶取代反應。微波輻射的老化過程中,首先發生Cu2+取代Zn5(CO3)2(OH)6中Zn2+生成(Cu,Zn)5(CO3)2(OH)6的同晶取代反應,并于1.0 h內基本完成;隨著老化時間繼續延長,主要進行Zn2+取代Cu2(CO3)(OH)2中Cu2+生成(Cu,Zn)2(CO3)(OH)2的同晶取代反應,同時(Cu,Zn)5(CO3)2(OH)6進一步結晶。與常規老化1 h制備的前驅體相比,微波輻射老化1.0 h制備的前驅體含有較多的(Cu,Zn)5(CO3)2(OH)6物相,有助于增強焙燒后CuO/ZnO/Al2O3催化劑中CuO-ZnO協同作用,提高表面銅含量,進而提高CuO/ZnO/Al2O3催化劑在漿態床合成甲醇的催化活性和穩定性,在400 h漿態床合成甲醇評價期間,甲醇時空收率最大達 318.9 g·kg-1·h-1,失活率僅為 0.11%·d-1。
微波輻射;甲醇;漿態床;CuO/ZnO/Al2O3催化劑
目前,工業甲醇的生產主要由CO催化加H2在固定床工藝中合成,其主要催化劑為CuO/ZnO/Al2O3催化劑[1-2]。CO加H2合成甲醇是一個強放熱反應,導致固定床反應器中催化劑床層易于過熱失活,所以生產中必須將原料氣的轉化率控制在較低水平,且采用高H2含量的合成氣,一般控制nH/nC比為4~7[3]。與固定床合成甲醇工藝相比,漿態床合成甲醇工藝具有反應溫度均勻,傳熱性好的特點,可以使用富CO合成氣,nH/nC比可達到化學計量,從而提高了原料氣的轉化率,但是催化劑的穩定性相對較低,工業化應用受到限制[4-14]。漿態床和固定床合成體系不同,對催化劑結構要求也不同[15-16],而催化劑前驅體的物相組成是影響催化劑結構的關鍵因素[17-20]。在催化劑前驅體的制備過程中,老化過程至關重要,影響催化劑前驅體晶相的生成和轉變,進而影響催化劑前驅體的物相組成[21-27]。Xia等[28]研究結果表明,在老化過程中,當物相由無定形向晶相轉變時,銅鋅開始摻入相應的堿式碳酸鹽,此時母液pH值降低;隨著銅鋅摻入量增加,逐步形成了大量能生成高活性的前驅體物相(Cu,Zn)2CO3(OH)2和(Cu,Zn)5(CO3)2(OH)6。Taylor等[29]研究表明,老化時間對CuO/ZnO/Al2O3催化劑中固溶體的形成影響較大,老化2.5 h時制備的催化劑活性較好。Li等[30]通過考察老化溫度,研究了CuO/ZnO/Al2O3催化劑前驅體晶相及組成變化對漿態床催化合成甲醇反應活性的影響,結果表明80℃老化制備的前驅體中主要為(Cu,Zn)2CO3(OH)2和(Cu,Zn)5(CO3)2(OH)6物相,焙燒后的催化劑比表面積大,CuO-ZnO協同作用強,在漿態床催化合成甲醇中的活性最高。
近年來,微波輻射技術在催化領域中的應用令人關注[31-32]。一般認為,微波輻射是介電加熱作用,即熱效應,并未改變反應動力學[33]。但最近的研究表明,微波輻射還存在非熱效應,能加快反應的速率,影響反應的選擇性,促進晶體的生長,控制材料的微觀結構[34]。Qi等[35]將微波輻射引入到甲烷芳構化 Mo/HZSM-5和Cu-Mo/HZSM-5催化劑的制備過程中,與常規加熱相比,微波輻射加熱制備催化劑的苯選擇性更高。Zhang等[36]對CuO/ZnO/Al2O3催化劑的前驅體進行3~10 min的微波輻射,使甲醇重整制氫的催化活性提高了7%。本工作將微波輻射引入CuO/ZnO/Al2O3催化劑制備的老化過程中,探討了不同微波輻射時間對CuO/ZnO/Al2O3前驅體晶相組成和催化劑微觀結構及其在漿態床合成甲醇工藝中性能的影響。
1 實驗部分
1.1 催化劑的制備
采用并流共沉淀法制備CuO/ZnO/Al2O3催化劑[16]。分別配制 1 mol·L-1Na2CO3(A.R.,天津市科密歐化學試劑有限公司)水溶液和Cu(NO3)2-Zn(NO3)2-Al(NO3)3(A.R.,天津市科密歐化學試劑有限公司)混合水溶液,于70℃水浴加熱并流加入到燒瓶中進行沉淀,Na2CO3水溶液流速為 3 mL·min-1,調節 Cu(NO3)2-Zn(NO3)2-Al(NO3)3混合溶液流速使沉淀液pH=7.5。然后將10%的沉淀母液(600 mL)置于微波化學反應器中(鞏義市予華儀器有限責任公司MCR-3,微波頻率2.45 GHz,微波最大輸出功率800 W,能夠自動調節功率以控溫)微波輻射控溫80℃老化不同的時間,然后抽濾,水洗,將濾餅于110℃干燥12 h獲得催化劑前驅體,再于350℃焙燒4 h,研磨過篩備用。催化劑表示為MWx,其中MW代表催化劑在微波輻射條件下老化,x表示老化時間,如MW1.0為在微波輻射條件下老化1.0 h制備的催化劑。常規80℃下老化 1.0 h 制備的催化劑標記為 NMW1.0。
1.2 催化劑反應性能評價
將5 g催化劑和250 mL液體石蠟(A.R.,天津市科密歐化學試劑有限公司)加入到500 mL高壓釜(WHF,威海自控反應釜有限公司)中,通入10%H2-90%N2的混合氣體進行程序升溫還原,于270℃還原6 h后切換為27%CO-68%H2-5%CO2原料氣,在240 ℃,4.0 MPa,氣體質量空速 1 620 L·(kg·h)-1條件下進行活性測試。反應后氣體經10℃水冷卻,減壓后經濕式流量計(LML-2,長春汽車濾清器有限責任公司)計量后排放,液相每12 h取樣進行稱量和分析。反應尾氣和液相產物均由GC9610型氣相色譜儀分析其組成。采用熱導池檢測器 (TCD)檢測,TDX201 色譜柱(3 cm×0.5 μm×2 m 不銹鋼柱)恒溫分析不凝性氣體產物,氬氣為載氣,流量20 mL·min-1,柱溫50℃,橋流60 mA,檢測器溫度和進樣口溫度均為200℃,進樣量1.0 mL,分流比60/1。用氫火焰離子化檢測器 (FID)檢測,PEG20M毛細管柱(0.32 mm×0.33 μm×30 m,石英毛細管柱)恒溫分析甲醇、乙醇、丙醇及烴類等液相產物組分,氮氣為載氣,流量 3.0 mL·min-1,H2流量 20 mL·min-1,空氣流量 200 mL·min-1,柱溫 150 ℃,進樣量 0.8 μL,分流比為60/1。樣品進樣口和檢測器溫度均為200℃,采用外標法定量計算。
1.3 催化劑的表征
催化劑X射線衍射(XRD)測試,在日本Rigaku D/max 2500型X射線衍射儀上進行,Cu Kα射線(經Kα2 剝離處理,λ=0.154056 nm),掃描速度 8°·min-1,石墨單色器,靶電壓和電流分別為40 kV和100 mA,步長 0.01°,掃描范圍 5°~85°,閃爍計數器記錄強度。
采用德國布魯克光譜儀器公司的VERTEX70型紅外光譜儀測定催化劑前驅體紅外光譜(FTIR),MCT檢測器,掃描范圍400~4 000 cm-1,掃描次數64,分辨率4 cm-1,掃描速度10 kHz,催化劑前驅體與KBr混合壓片制樣。
采用德國耐馳公司STA409C型熱分析儀進行熱重-微分熱分解(TG-DTG)實驗,樣品質量約30 mg,N2流量 40 mL·min-1,O2流量 10 mL·min-1,升溫速率8℃·min-1。程序升溫還原(H2-TPR)實驗在美國Micromeritics公司AurochemⅡ2920型全自動程序升溫化學吸附儀上進行,催化劑用量約20 mg,置于U形石英反應管中,He氣流量50 mL·min-1,以10℃·min-1升至300℃,恒溫吹掃30 min,然后降溫至40℃,切換為10%H2-90%Ar混合氣,流量50 mL·min-1。待系統穩定后,以 10℃·min-1升至600℃,TCD檢測H2消耗量。
HR-TEM分析采用GG314-JEM-2100F場發射透射電子顯微鏡,加速電壓200 kV,將催化劑樣品分散在乙醇溶液,并通過超聲波超聲10 min,將懸浮液滴在通網上制樣。
X射線光電子能譜 (XPS)測定采用英國VG Scientific公司ESCAL-ab 220i-XL型光電子能譜儀,激發源為Al Kα X射線,功率約為300 W。分析時的基礎真空為3×10-4Pa,電子結合能用污染碳的C1s峰(284.6 eV)作為內標校正。
2 結果與討論
2.1 催化劑活性及穩定性測試
在微波輻射條件下,不同老化時間制備的催化劑在漿態床合成甲醇工藝中的評價結果見圖1。由圖可見,在微波輻射條件下,隨著老化時間的延長,催化劑的活性和穩定性先升高后降低。老化0.5 h制備的 MW0.5 催化劑的活性低于 275 g·kg-1·h-1,且失活較快。而老化1.0 h制備的MW1.0催化劑活性最好,甲醇時空收率STY最高達到320 g·kg-1·h-1,失活也較慢。老化1.5 h制備的MW1.5催化劑活性略有下降,但是催化劑穩定性更好。當老化時間延長至2.0和2.5 h時,催化劑活性和穩定性急劇下降。和常規老化1.0 h制備的催化劑NMW1.0相比,微波輻射老化1 h制備的催化劑MW1.0的活性和穩定性顯著提高。
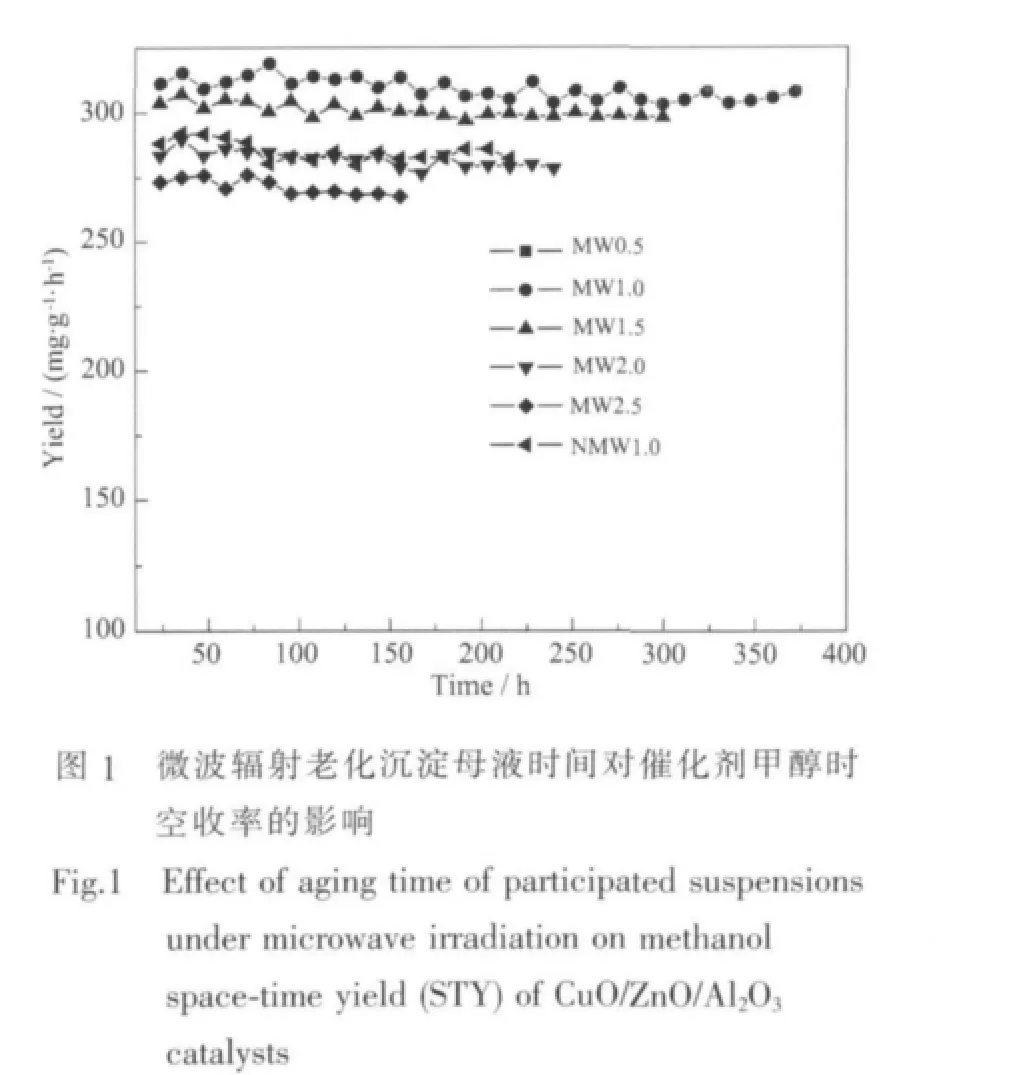
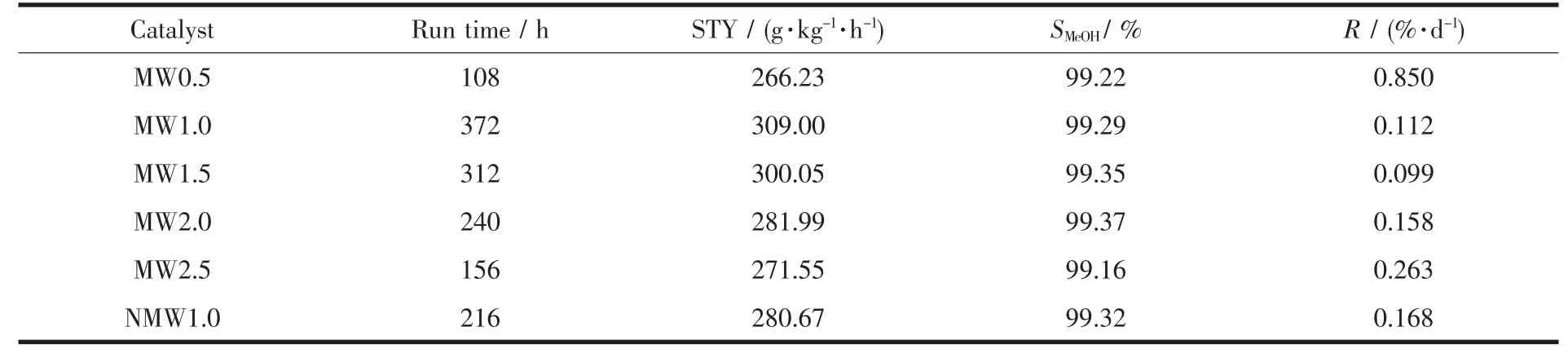
表1 催化劑在漿態床中的活性評價平均數據Table 1 Average catalysis data of catalysts evaluated in the slurry methanol synthesis
表1是不同催化劑的活性評價期間的甲醇平均時空收率STY,選擇性SMeOH和失活率R。由表可知,所有催化劑的甲醇選擇性差別較小,均高于99%。MW1.0和 MW1.5的平均甲醇時空收率 STY與NMW80 相比分別提高了 10.1%和 6.9%,且 MW1.0,MW1.5 和 MW2.0 的失活率明顯低于 NMW1.0 催化劑。可見,在催化劑的老化過程中,微波輻射有助于提高漿態床催化劑的活性和穩定性,尤其對催化劑穩定性的提高更加明顯。
2.2 催化劑前驅體XRD表征
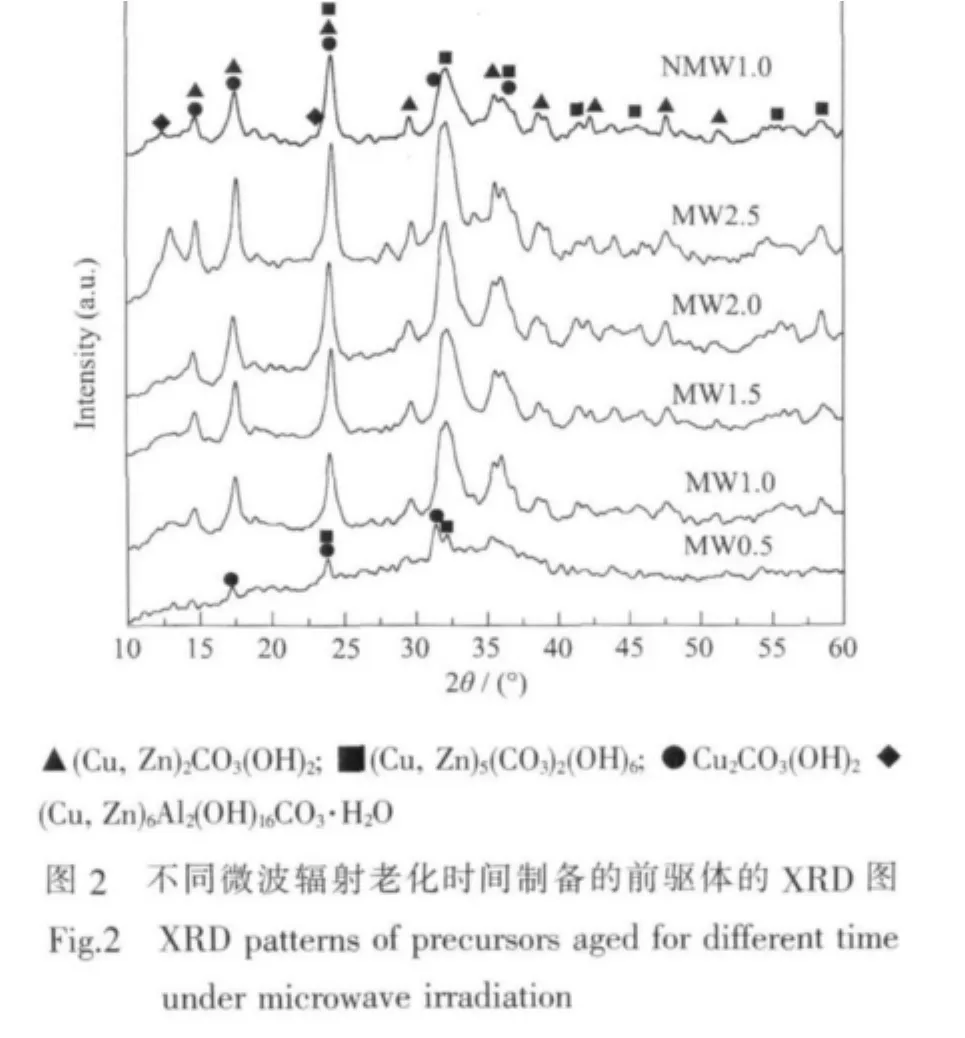
圖2為不同老化條件下催化劑前軀體的XRD圖。由圖可知,MW0.5 前軀體在 2θ=17.2°,23.8°,32°和 32.15°出現較弱的衍射峰,其中 2θ=23.8°和 32°分別為 Cu2CO3(OH)2(JCPDS41-1390)和 (Cu,Zn)5(CO3)2(OH)6(JCPDS38-0152)的獨立特征衍射峰,可知,前驅體中出現Cu2CO3(OH)2和(Cu,Zn)5(CO3)2(OH)6晶相,說明在微波輻射條件下老化0.5 h時,Cu2+開始取代Zn5(CO3)2(OH)6晶格中 Zn2+形成(Cu,Zn)5(CO3)2(OH)6晶相。當老化時間延長至1 h時,(Cu,Zn)5(CO3)2(OH)6晶相的衍射峰增大,且在 2θ=36.0°,41.4°,45.9°,55.1°和 58.4°出現該晶相的其它衍射峰,同時在 2θ=29.6°,35.5°,38.6°,42.3°,47.7°和 51.1°出現鋅孔雀石(Cu,Zn)2CO3(OH)2(JCPDS36-1475)的晶相衍射峰,表明在老化時間為0.5~1.0 h的過程中,仍不斷進行Cu2+取代 Zn5(CO3)2(OH)6晶格中 Zn2+形成(Cu,Zn)5(CO3)2(OH)6的同晶取代反應,同時Zn2+開始取代Cu2CO3(OH)2中的 Cu2+形成(Cu,Zn)2CO3(OH)2晶相。隨著老化時間的繼續延長,前驅體的XRD衍射峰逐漸增強,可知(Cu,Zn)5(CO3)2(OH)6和(Cu,Zn)2CO3(OH)2的結晶度逐漸增大。當老化2.5 h時,前軀體在2θ=11.8°出現一新衍射峰,歸屬為類水滑石(Cu,Zn)6Al2(OH)16CO3·4H2O 晶相[37](JCPDS14-191)。對比 NMW1.0前軀體與MW1.0前驅體的XRD圖可知,兩前驅體具有相同的特征衍射峰,但NMW1.0前驅體的(Cu,Zn)5(CO3)2(OH)6的衍射峰較矮較寬泛,而MW1.0前驅體中(Cu,Zn)5(CO3)2(OH)6的衍射峰更加尖銳,可知微波輻射更有助于物相(Cu,Zn)5(CO3)2(OH)6的結晶。
2.3 前驅體FTIR表征
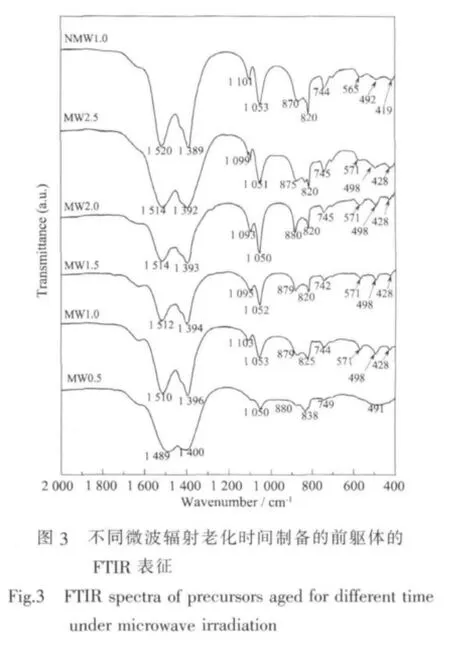
圖3為微波輻射老化和常規老化制備前驅體的FTIR吸收光譜圖。由圖可知,微波老化0.5 h制備的前驅體在1 489、1 400、1 050和838 cm-1處出現孔雀石Cu2(CO3)(OH)2/Zn5(CO3)2(OH)6結構的C-O傅里葉紅外特征吸收峰[30]。在微波輻射條件下,隨著老化時間的延長,各前驅體在1530~1380 cm-1波段內的C-O不對稱伸縮振動吸收峰都較MW0.5前驅體發生偏移,1 489 cm-1處吸收峰逐漸向1 514 cm-1移動,1400 cm-1處吸收峰向1392 cm-1移動,發生這種現象的原因是Zn2+取代了Cu2(CO3)(OH)2中的Cu2+,導致C-O不對稱伸縮振動吸收峰的高波數發生藍移、低波數發生紅移,且隨著Zn2+取代量的增多,低波數發生紅移、高波數發生藍移的程度越大[38]。可知在微波輻射條件下,隨著老化時間的延長,不斷有Zn2+取代Cu2(CO3)(OH)2中的Cu2+生成更多的(Cu,Zn)(CO3)(OH)2物相。NMW1.0的C-O 不對稱伸縮振動吸收峰為1520和1389 cm-1,高波段藍移,低波段紅移的程度更大,說明NMW1.0前軀體中較 MW1.0,MW1.5,MW2.0 和 MW2.5 中含有更多的Cu2(CO3)(OH)2物相。
綠銅鋅礦在CO32-的傅里葉紅外特征吸收波段和鋅孔雀石的發生重疊,區別不明顯,但在600~400 cm-1的Me-O(金屬氧鍵)傅里葉紅外特征吸收波段,由于Cu-O的相互作用比Zn-O的強,所以當Zn5(CO3)2(OH)6中的Zn2+被Cu2+取代后,Me-O傅里葉紅外特征吸收波段的吸收峰變強且都發生藍移,同時隨著Cu2+取代量的增多,Me-O吸收峰藍移的程度越大[38]。MW0.5 前驅體在 600~400 cm-1范圍內僅在491 cm-1波段出現吸收峰。當老化時間為1.0 h時,前驅體在491 cm-1處的吸收峰藍移至498 cm-1,同時在571和428 cm-1出現2個新的吸收峰。隨著老化時間的繼續延長,前驅體在600~400 cm-1范圍內的吸收峰都為498,571和428 cm-1,不再發生偏移,表明在微波輻射條件下,老化1 h就基本完成了Cu2+取代Zn5(CO3)2(OH)6中的Zn2+的同晶取代過程。在無微波輻射條件下老化1.0 h制備的NMW1.0前驅體 在 600~400 cm-1的吸收 峰為 565,492和419 cm-1,均低于在微波輻射條件下老化制備的前驅體 MW1.0,表明 MW1.0 前驅體比 NMW1.0 含有更多的綠銅鋅礦物相(Cu,Zn)5(CO3)2(OH)6。
2.4 前驅體DTG表征
圖4為微波輻射條件下催化劑前驅體的DTG圖,由圖可知,MW0.5前驅體在150~350℃出現寬泛的失重峰,在364℃出現了一個較小失重峰,第一個失重峰是由Cu2CO3(OH)2和無定形的(Cu,Zn)(CO3)(OH)2混合物分解產生的,見反應式(1),由于晶體中Cu2+/Zn2+摻入的混雜性,使晶體穩定性較差,導致兩物相的分解溫度較低且失重峰較寬泛[27-28,30],第二個失重峰為綠銅鋅礦(Cu,Zn)5(CO3)2(OH)6物相分解產生,見反應式(2),且在455℃出現了碳酸鹽的失重峰[24],見反應式(3)和(4)。與MW0.5 前驅體的 DTG 曲線相比,MW1.0 在 288 ℃出現較明顯的(Cu,Zn)2(CO3)(OH)2失重峰,且綠銅鋅礦(Cu,Zn)5(CO3)2(OH)6失重峰變大,同時碳酸鹽的分解溫度略有升高,結合前驅體的XRD和FTIR表征結果可知,這是由于MW1.0前驅體中出現有序的 (Cu,Zn)(CO3)(OH)2晶相,同時(Cu,Zn)5(CO3)2(OH)6的含量和結晶度增大造成的。MW1.5,MW2.0 和 MW2.5 前驅體的失重曲線與MW1.0的相似,各失重峰都向高溫方向移動,表明各物相的結晶度逐漸增大。常規老化的NMW1.0前驅體在400℃以下出現一寬泛的失重峰,由前驅體的XRD和FTIR表征可知,該前驅體中(Cu,Zn)2(CO3)(OH)2含量較大,(Cu,Zn)5(CO3)2(OH)6含量較小,且結晶度較低,所以(Cu,Zn)5(CO3)2(OH)6失重溫度較低,失重峰較寬泛,且與(Cu,Zn)2(CO3)(OH)2的失重曲線發生重疊,從而出現一寬泛的失重峰。結合TG數據,進一步將各前驅體中鋅孔雀石(Cu,Zn)2(CO3)(OH)2物相和綠銅鋅礦(Cu,Zn)5(CO3)2(OH)6物相的失重峰溫和失重量總結于表2。
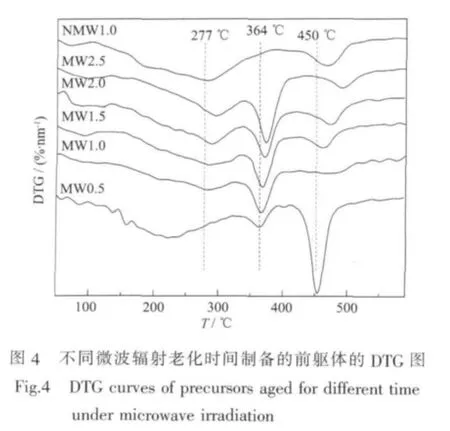
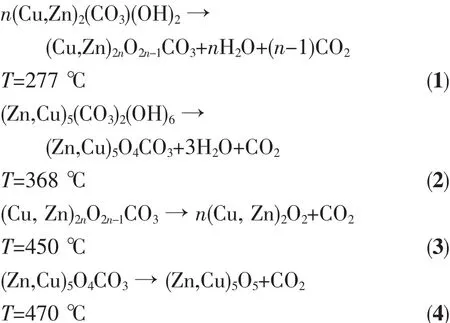
由表2可知,在微波輻射條件下,老化0.5 h制備的前驅體中綠銅鋅礦的失重量較小。當老化1 h,出現鋅孔雀石的失重,且前驅體中綠銅鋅礦失重量增大,同時兩物相的失重溫度升高。隨著老化時間進一步的延長,前驅體中鋅孔雀石的失重量逐漸增大,而綠銅鋅礦的失重量基本不變,同時兩物相的失重溫度逐漸升高,表明在微波輻射的整個老化過程中不斷有鋅孔雀石生成,但當老化1.0 h以后不再有綠銅鋅礦物相生成,且兩物相的結晶度都隨著老化溫度的升高逐漸變大,這與前驅體的FTIR表征結果相符。無微波輻射制備的NMW1.0前驅體中鋅孔雀石物相的含量較高,而綠銅鋅礦含量較少,再次說明,微波輻射有助于綠銅鋅礦物相的生成。
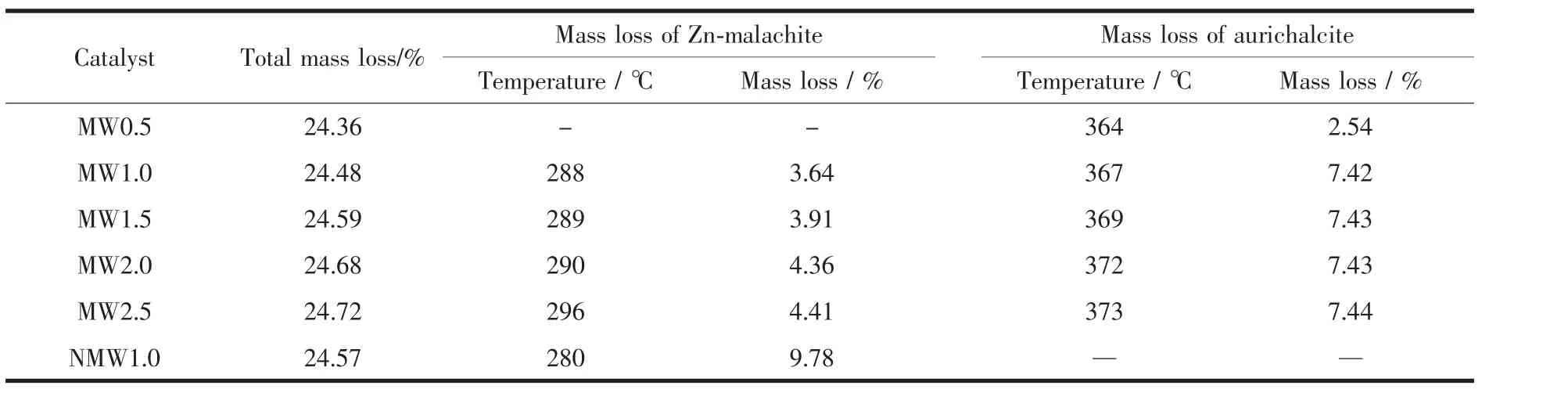
表2 不同時間微波輻射老化的前驅體失重分析數據Table 2 Mass loss analytic data of precursors aged for different time under microwave irradiation
在并流沉淀過程中,由于溶液一直保持較高的過飽和度,沉淀過程中主要生成無定形的Cu2(CO3)(OH)2,Zn5(CO3)2(OH)6物相,如(5)~(8)反應。而老化過程主要是進行同晶取代反應,即Cu2+進入Zn5(CO3)2(OH)6晶格中取代 Zn2+形成(Cu,Zn)5(CO3)2(OH)6物相和 Zn2+同晶取代 Cu2CO3(OH)2中的 Cu2+形成(Cu,Zn)2CO3(OH)2物相的過程[27-28,39],如式(9)和(10)。
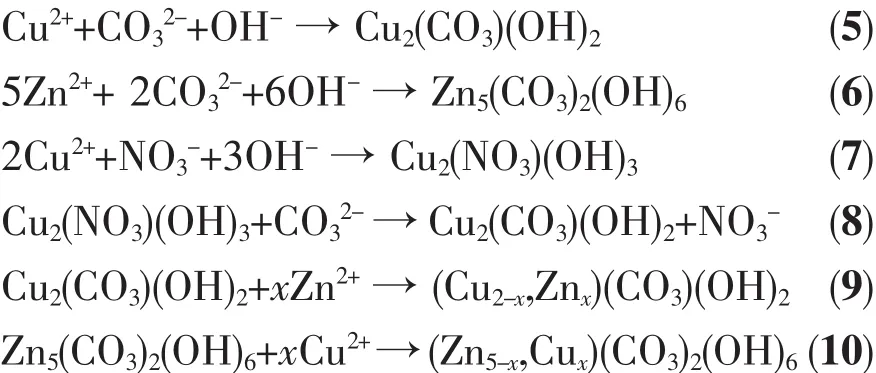
結合上述前驅體的XRD,FTIR和DTG表征結果可知,在微波輻射條件下,當老化時間為0.5 h時,有少量的(Cu,Zn)5(CO3)2(OH)6晶相的形成;當老化時間延長至1.0 h,完成Cu2+取代Zn5(CO3)2(OH)6中的Zn2+的過程,同時前驅體中(Cu,Zn)2(CO3)(OH)2物相的含量增大;隨著老化時間的進一步延長,仍不斷有(Cu,Zn)2(CO3)(OH)2生成。結合在老化時間都為1 h 條件下,MW1.0 前驅體比 NMW1.0 前驅體中含有更多的綠銅鋅礦(Cu,Zn)5(CO3)2(OH)6物相這一結論。我們可以進一步得出,在沉淀母液的老化過程中,微波輻射可以促進Cu2+進入Zn5(CO3)2(OH)6晶格中取代Zn2+(反應式6)的優先進行。而NMW1.0前驅體中(Cu,Zn)5(CO3)2(OH)6含量較少的原因是,原料中nCu∶nZn=2∶1,原料液中 Cu2+濃度較大,Zn2+濃度較小,所以在沉淀初期生成較多的Cu2(CO3)2(OH)2物相,使沉淀母液中Cu2(CO3)(OH)2濃度較大,反應物濃度是影響反應的進行主要因素,所以Zn2+取代Cu2CO3(OH)2中 Cu2+的為主要反應(反應式 5),導致 NMW1.0前驅體中鋅孔雀石(Cu,Zn)2(CO3)(OH)2物相的含量較大,而綠銅鋅礦(Cu,Zn)5(CO3)2(OH)6物相的含量較少。
2.5 催化劑的XRD表征
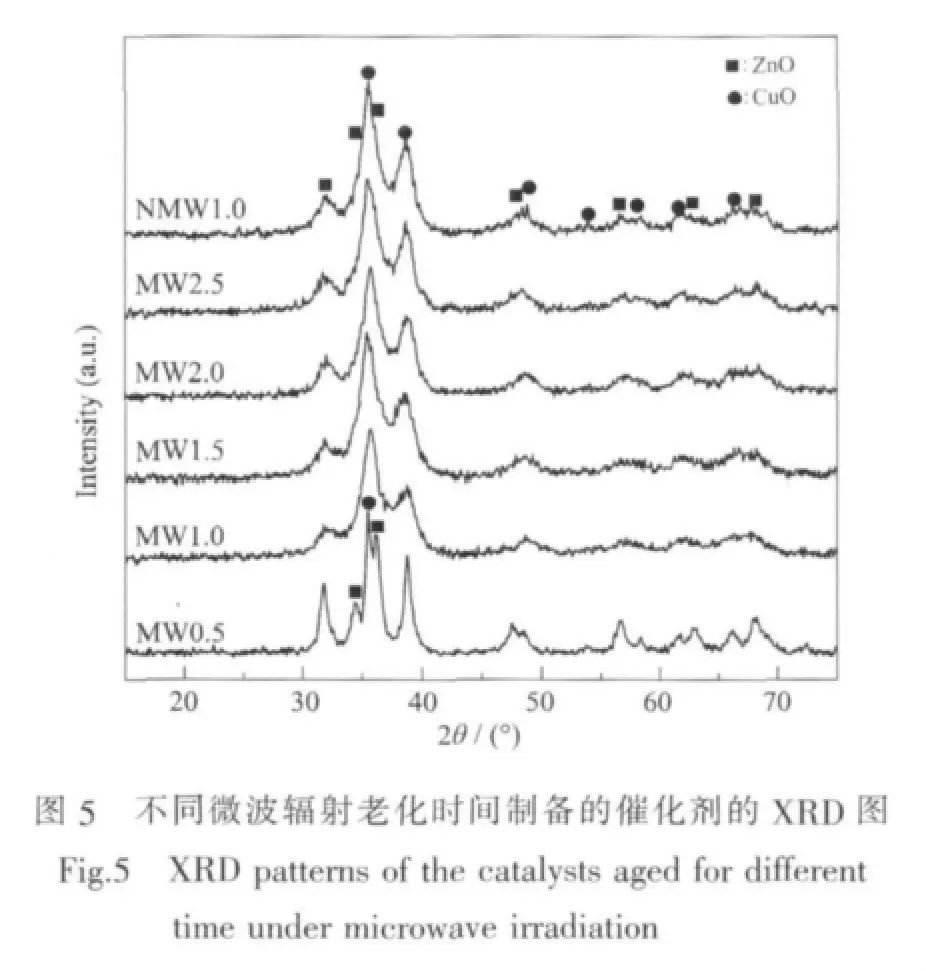
圖5為不同老化條件下制備的催化劑XRD圖,并利用催化劑中CuO在2θ≈38.7°衍射峰進行高斯擬合,通過半峰寬用Scherrer公式計算CuO平均粒徑,其結果見表 4。由圖可知,各催化劑在 2θ=35.4°和38.7°處均出現明顯的CuO的XRD特征峰,且在2θ=31.9°,34.4°和 36.4°處出現 ZnO 的主要特征衍射峰,但沒有檢測到Al2O3相,可知催化劑中的Al2O3以無定形的狀態存在[16]。在微波輻射條件下,MW0.5催化劑的CuO和ZnO的衍射峰較尖銳且相互分離,這是由于微波輻射老化0.5 h時,沒有完成Zn2+/Cu2+取代 Cu2CO3(OH)2/Zn5(CO3)2(OH)6中Cu2+/Zn2+過程,使前驅體分解后形成分離的CuO和ZnO,且CuO和ZnO 晶粒較大(≈6.11 nm)。當老化1 h時,催化劑的XRD 衍射峰變矮變寬,且在 2θ=38.7°的 CuO 與 2θ=34.4°和 36.4°的 ZnO 的衍射峰相互重疊,這是由于老化1 h完成Cu2+取代Zn5(CO3)2(OH)6中Zn2+的過程,同時有更多的 (Cu,Zn)2(CO3)(OH)2物相生成,MW1.0前驅體焙燒后形成CuO-ZnO固溶體,催化劑中的CuO和ZnO分散程度高[40],CuO晶粒變小,粒徑僅為3.50 nm,使該催化劑活性最高。隨著老化時間的進一步延長,雖然(Cu,Zn)2(CO3)(OH)2物相含量略有增加,但(Cu,Zn)5(CO3)2(OH)6和(Cu,Zn)2(CO3)(OH)2物相的結晶度逐步增大,分解后的CuO晶粒逐漸變大(表3),衍射峰逐漸增強[28],催化劑活性和穩定性逐漸下降。當老化時間為均1 h時,與常規老化的催化劑NMW1.0相比,微波輻射老化的催化劑MW1.0中 CuO和 ZnO衍射峰更矮更寬泛,且MW1.0催化劑中CuO晶粒的粒徑(3.5 nm)遠小于NMW1.0 中的(6.2 nm),結合前驅體表征分析可知,這是由于MW1.0前驅體中含有更多的綠銅鋅礦物相,而綠銅鋅礦為單斜晶系,氧原子以雙層緊密堆積方式排列,Cu2+在八面體的中心,而Zn2+位于四面體位置上,分解生成的CuO-ZnO固溶體可保證CuO充分細化,ZnO分散在CuO的周圍,防止CuO的團聚[41-42],進而提高催化劑的活性和穩定性。
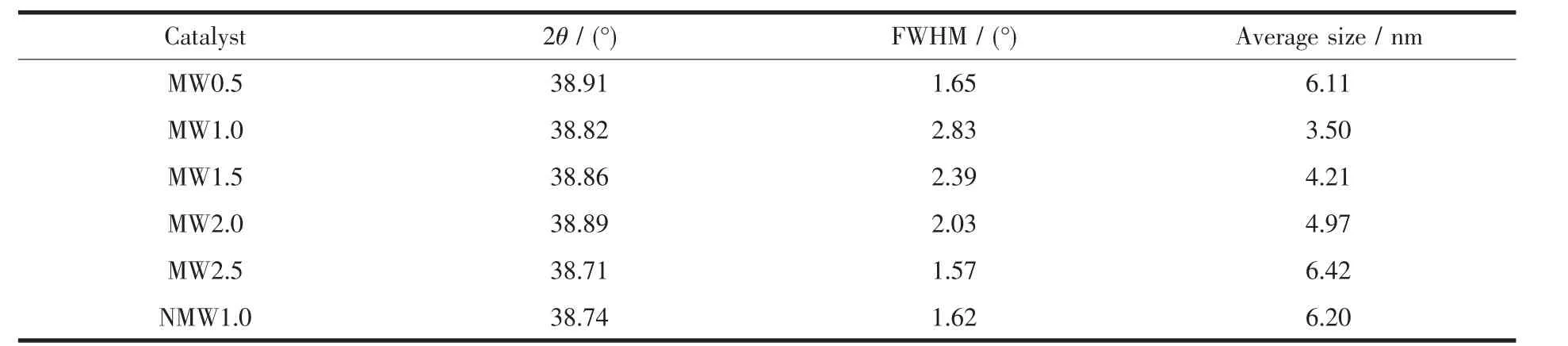
表3 催化劑中CuO(2θ≈38.84°)晶粒的分析結果Table 3 Analytic results of the CuO crystal(2θ≈38.84°)in the catalysts
2.6 催化劑的HR-TEM表征
圖6為燒后催化劑 MW1.0、MW1.5 和 NMW1.0的HR-TEM圖。從圖中可以看出,在微波輻射條件下老化1.0 h制備的催化劑MW1.0中,CuO的晶粒較小,粒徑在3~4 nm之間,這與Scherrer公式計算的結果相似,當老化時間延長至1.5 h時,催化劑中CuO晶粒變大,粒徑在4~5 nm之間,無微波輻射老化制備的催化NMW1.0的CuO晶粒最大,在6 nm左右。進一步對比可知,在微波輻射條件下老化制備的催化劑CuO晶粒被ZnO包圍,CuO晶粒分散程度高,而無微波老化制備的催化劑中,CuO晶粒自身發生團聚,分散程度差。
2.7 催化劑的H2-TPR表征
將微波條件下制備的前驅體進行焙燒,對所得到的催化劑進行H2-TPR表征,發現各催化劑均有兩個還原峰,即低溫還原峰(PeakⅠ)和高溫還原峰(PeakⅡ),進一步通過高斯擬合對還原峰進行分峰,結果見圖7。在微波輻射條件下,隨著老化時間的延長,催化劑的還原溫度先降低后升高,且催化劑的低溫峰(PeakⅠ)面積始終大于高溫峰(PeakⅡ)。而對于無微波輻射老化的催化劑,高溫還原峰的面積卻遠大于低溫還原峰。兩個還原峰均歸屬于Cu2+還原為金屬銅的過程,低溫還原峰對應于與ZnO發生強相互作用或高分散的CuO的還原,高溫還原峰對應大晶粒體相銅的CuO的還原[15,43]。
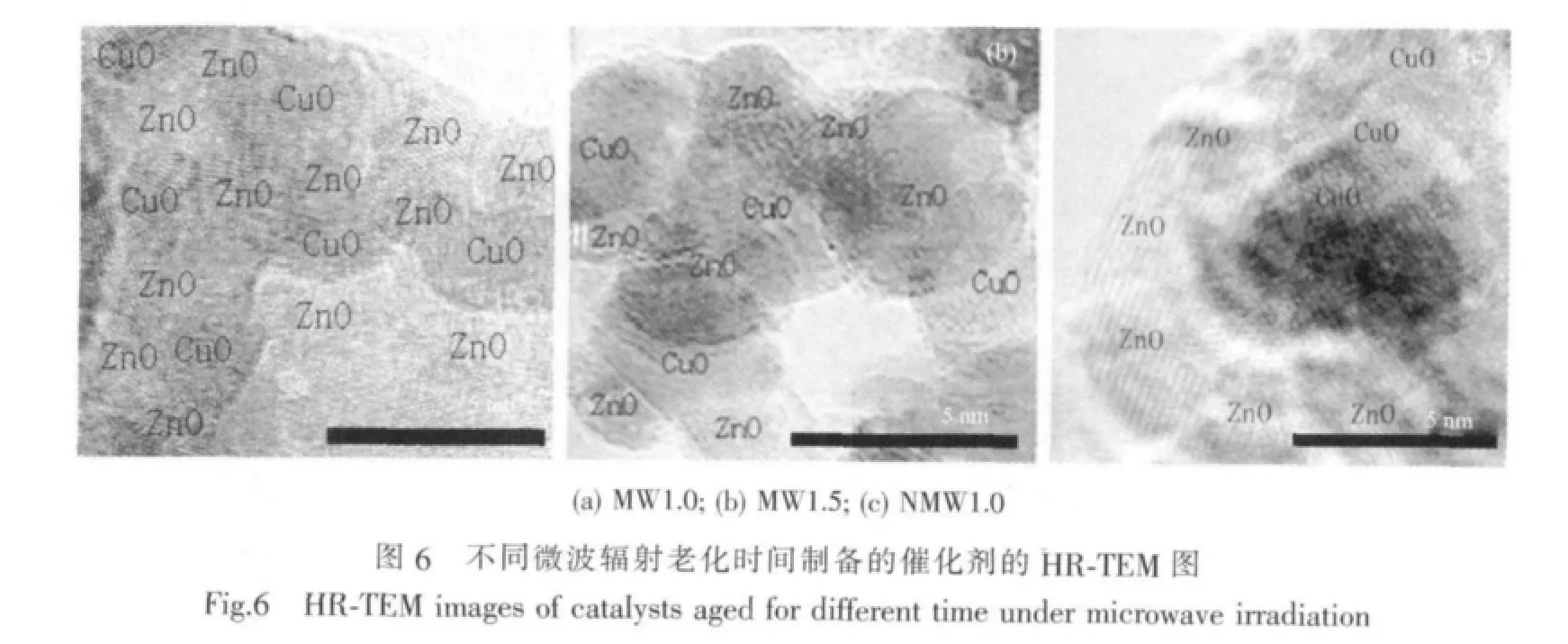
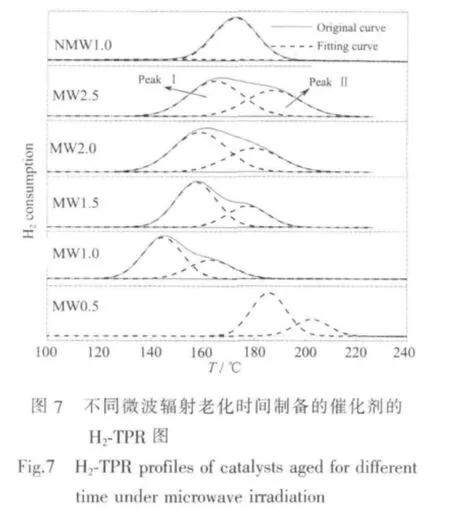
表3為催化劑的H2-TPR的高斯擬合分峰數據。由表可知,MW0.5催化劑還原溫度最高,分別為186和203℃,結合前軀體的表征可知,MW0.5沒有完成 Zn2+/Cu2+取代 Cu2CO3(OH)2/Zn5(CO3)2(OH)6中Cu2+/Zn2+的過程,所以可將低溫還原峰歸于Cu2CO3(OH)2分解的CuO的還原,高溫還原峰歸于(Cu,Zn)5(CO3)2(OH)6和(Cu,Zn)2(CO3)(OH)2分解的 CuO 的還原。在微波輻射條件下,隨著老化時間繼續延長,MW1.0、MW1.5、MW2.0 和 MW2.5 催化劑的低溫還原峰 (Peak I)的相對面積逐漸減小,高溫還原峰(Peak II)的相對面積逐漸增大,其變化規律恰好與表2分析的前驅體中 (Cu,Zn)5(CO3)2(OH)6和(Cu,Zn)2(CO3)(OH)2物相含量的變化規律一致。同時據Reddy等[44]的研究可知,(Cu,Zn)5(CO3)2(OH)6物相分解后的CuO和ZnO達到原子級混合,相互作用較強,而(Cu,Zn)2(CO3)(OH)2分解后的CuO和ZnO以微小顆粒形式混合,相互作用較弱。可進一步將低溫還原峰對應于(Cu,Zn)5(CO3)2(OH)6物相的分解的CuO還原,高溫還原峰對應于(Cu,Zn)2(CO3)(OH)2物相的分解的CuO的還原。雖然 NMW1.0前驅體含有(Cu,Zn)5(CO3)2(OH)6和(Cu,Zn)2(CO3)(OH)2兩物相,但由于綠銅鋅礦(Cu,Zn)5(CO3)2(OH)6含量較少,且結晶度較低,導致其分解之后的CuO晶粒較大,燒后催化劑的CuO還原溫度較高,與(Cu,Zn)2(CO3)(OH)2分解之后的CuO的還原峰重疊。
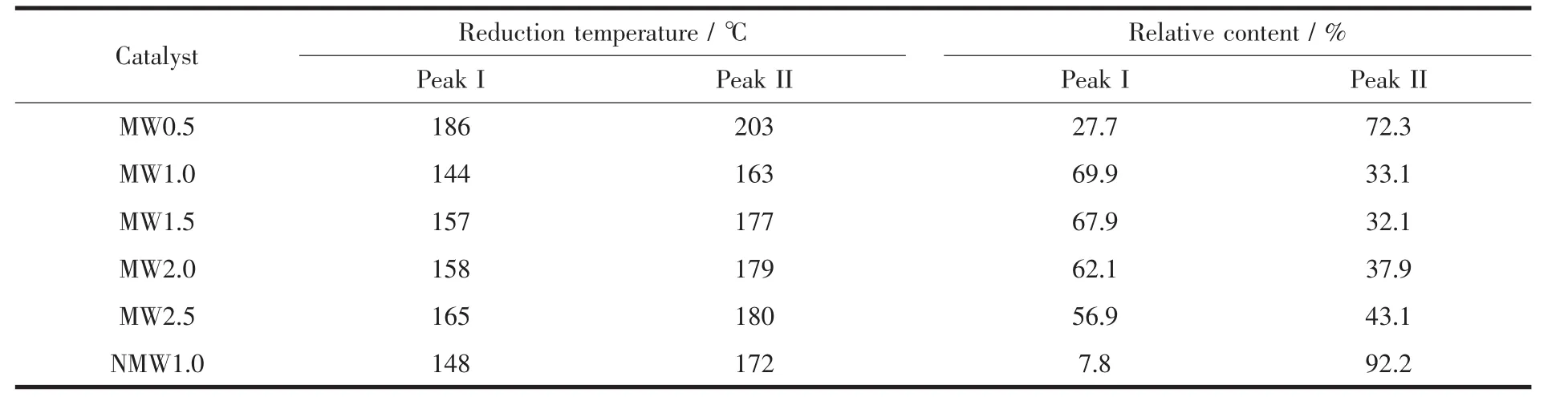
表4 不同微波輻射老化時間制備的催化劑H2-TPR分析結果Table 4 H2-TPR analytic results of the catalysts aged for different time under microwave irradiation
2.8 催化劑的XPS表征
焙燒后催化劑的Cu2p3/2的XPS譜如圖8(a)所示。由圖可見,催化劑的Cu2p3/2結合能均在932~934 eV,同時在高位端940~945 eV有強衛星半峰出現,說明Cu在催化劑表面以Cu2+形式存在[15,30]。在微波輻射條件下,隨著老化時間的延長催化劑的Cu2p3/2結合能分別為 933.02,933.96,933.75,933.47和933.12 eV,先升高后降低;而由圖8(b)可知,它們的 Zn2p3/2結合能分別為1022.47,1 021.06,1 021.60,1 02184,1 022.29 eV,先降低后升高,說明催化劑中Cu和Zn組分所處的化學環境和能量狀態發生了改變,發生結合能偏移的原因是CuO-ZnO協同作用,這是由于Zn的電負性比Cu高,Cu最外層電子向Zn偏移,從而使Cu的電子云密度減小,電子結合能增加,Zn的電子云密度增加,電子結合能減小[17]。表明在微波輻射條件下,隨著老化時間的延長,催化劑中CuO-ZnO的協同作用先變強后變弱,其中MW1.0中CuO-ZnO的協同作用最強。而焙燒后催化劑NMW1.0中Cu2p3/2和Zn2p3/2結合能分別為 933.31 和 1 022.05 eV,Cu2p3/2和 Zn2p3/2結合能的偏移程度都小于MW1.0,表明催化劑MW1.0中CuO-ZnO作用強于NMW1.0。
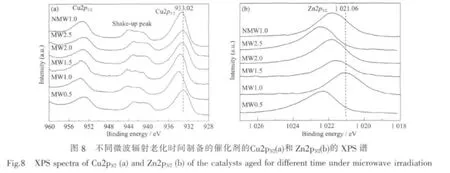
表5為焙燒后CuO/ZnO/Al2O3催化劑的表面原子含量。由表可知,在微波輻射條件下,隨著老化時間的延長,催化劑的表面Cu/Zn先升高后降低,其中MW1.0的表明銅含量最高,且遠高于無微波輻射老化制備的催化劑NMW1.0。表面銅含量也是影響催化劑活性的重要因素,表面銅含量越高催化劑的活性越高,反之催化劑的活性越低[15],此結論也與催化劑的活性評價結果相一致。由催化劑前驅體的表征結果可知,前驅體中(Cu,Zn)5(CO3)2(OH)6物相的含量與催化劑表面銅含量正相關,表明綠銅鋅礦的存在有利于催化劑表面Cu含量的提高,進而提高催化性能。
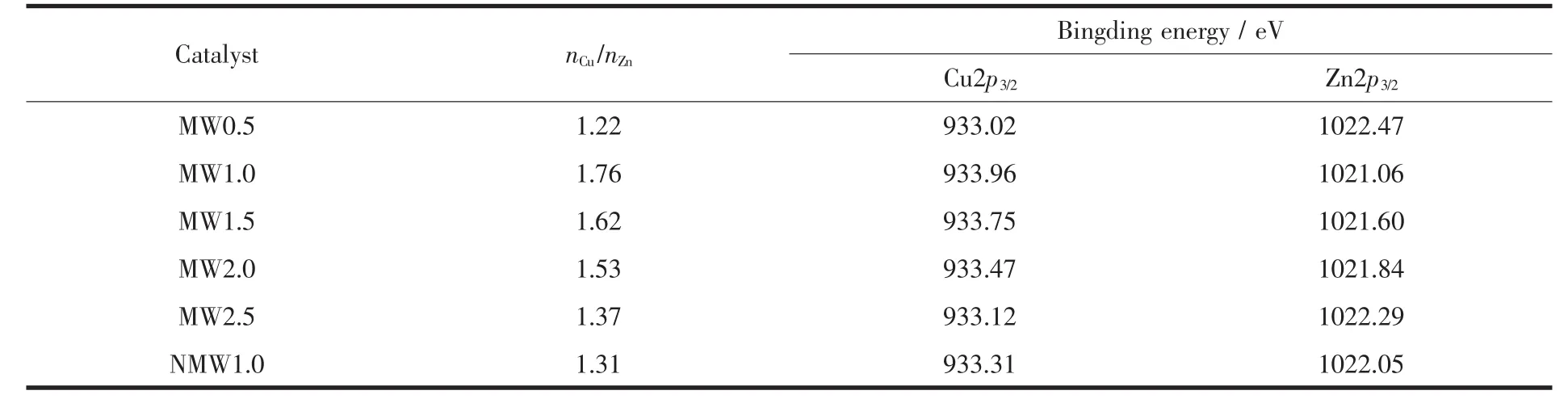
表5 不同微波輻射老化時間制備的催化劑的XPS數據分析Table 5 XPS analytic data of the catalysts aged for different time under microwave irradiation
3 結 論
在催化劑的老化過程中,微波輻射有助于Cu2+取代Zn5(CO3)2(OH)6中的Zn2+的過程,提高了前驅體中綠銅鋅礦物相的含量,焙燒后催化劑的CuO-ZnO協同作用增強,CuO晶粒變小,表面銅含量增高,大大提高Cu/ZnO/Al2O3催化劑在漿態床工藝中的活性和穩定性。在微波輻射條件下,老化1 h制備的催化劑的活性和穩定性最好,在400 h漿態床合成甲醇評價期間,時空收率最大為 318.9 g·kg-1·h-1,平均失活率僅為0.11%·d-1,比非微波輻射制備的催化劑的甲醇時空收率提高了10.1%,平均失活率降低了31.2%。
[1]Chiotti G,Boccuzzi F.Cat.Rev.Sci.Eng.,1987,29:151-233
[2]Chinchen G C,Denny P J,Jennings J R,et al.Appl.Catal.A.,1988,36:1-65
[3]DIAO Xue-Ying(刁雪瑩),ZHANG Ji-Bo(張吉波),LI Guang-Wei(李廣偉),et al.Sci.&Technol.Chem.Ind.(Huagong Keji),2000,8(6):49-52
[4]Tijm P J A,Waller F J,Brown D M.Appl.Catal.A,2001,221(8):275-282
[5]Cybulski A.Cat.Rev.Sci.Eng.,1994,36(4):557
[6]Wender I.Fuel Process.Technol.,1996,48(3):189-297
[7]Wang J H,Anthony R G,Akgerman A.Comput.Chem.Eng.,2005,29(11/12):2474-2484
[8]Setinc M,Levec J.Chem.Eng.Sci.,1999,54(15/16):3577-3586
[9]Lee S,Sardesai A.Top.Catal.,2005,32(3/4):197-207
[10]Zhai X F,Shamoto J,Xie H J,et al.Fuel,2008,87(3/4):430-434
[11]Air Products and Chemicals.Inc.Final Report.June 2003.
[12]Quinn R,Mebrahtu T,Dahl T A,et al.Appl.Catal.A,2004,264(1):103-109
[13]Twigg M V,Spencer M S.Appl.Catal.A,2001,212(1/2):161-174
[14]Van der Laan G P,Beenackers A A C M,Ding B Q,et al.Catal.Today,1999,48(1/2/3/4):83-92
[15]LI Zhong(李忠),ZHENG Hua-Yan(鄭華艷),XIE Ke-Chang(謝克昌).Chin.J.Catal.(CuihuaXuebao),2008,29(05):431-435
[16]Zhang X B,Li Z,Guo Q H,et al.Fuel,2010,91(4):379-382
[17]CAO Yong(曹勇),CHEN Li-Fang(陳立芳),DAI Wei-Lin(戴維林),et al.Chem.J.Chinese Universities(Gaodeng Xuexiao Huaxuex Xebao),2003,24(07):1296-1298
[18]HONG Zhong-Shan(洪中山),DENG Jing-Fa(鄧景發),FAN Kang-Nian(范康年),et al.Chem.J.Chinese Universities(Gaodeng Xuexiao Huaxuex Xebao),2002,23(04):706-708
[19]Jensen J R,Johannessen T,Wedel S,et al.J.Catal.,2003,218(1):67-77
[20]XU Hui-Yuan(徐慧遠),CHU Wei(儲偉),CI Zhi-Min(慈志敏).Acta Phys.-Chem.Sin.(Wuli Huaxue Xuebao),2007,23(7):1042-1046
[21]ZHANG Xi-Tong(張喜通),CHANG Jie(常杰),WANG Tie-Jun(王鐵軍),et al.J.Fuel.Chem.Technol.(Ranliao Huaxue Xuebao).2005,33(4):479-482
[22]Baltes C,Vukojevic S,Schuth F.J.Catal.,2008,258(2):334-344
[23]Bems B,Schur M,Dassenoy A,et al.J.Chem.Educ.,2003,9(9):2039-2052
[24]Millar G J,Holm I H,Uwins P J R,et al.J.Chem.Soc.Faraday Trans.,1998,94(4):593-600
[25]Muhamad E N,Irmawati R,Taufiq-Yap Y H,et al.Catal.Today,2008,131(1/2/3/4):118-124
[26]Porta P,De R S,Ferraris G.J.Catal,1988,109(2):367-377
[27]Li J L,Inui T.Appl.Catal.A,1996,137(1):105-117
[28]XIA Wang-Qiong(夏王瓊),TANG Hao-Dong(唐浩東),LIN Sheng-Da(林盛達),et al.Chin.J.Catal.(Cuihua Xuebao),2009,30(9):879-884
[29]Taylor S H,Hutchings G J,Palacio M L.et al.Catal.Today,2003,84(3/4):113-119
[30]LI Zhong(李忠),GUO Qi-Hai(郭啟海),ZHANG Xiao-Bing(張小兵),et al.Chem.J.Chinese Universities(Gaodeng Xuexiao Huaxuex Xebao),2009,30(11):2215-2221
[31]Ren J,Liu S S,Li Z,et al.Appl.Catal.A,2009,366(1):93-101
[32]Lingaiah N,Sai Prasad P S,Kanta R P,et al.Catal.Commun.,2002,3(9):391-397
[33]Baghurst D R,Mingos D M P.J.Organomet.Chem.,1990,384(3):C57-C60
[34]ZHU Rui-Zhi(朱睿智),MENG Xian-Liang(孟獻梁),ZONG Zhi-Min(宗志敏).Mod.Chem.Ind.(Xiandai Huagong),2007,27(1):383-386
[35]Qi S T,Yang B L.Catal.Today,2004,98(4):639-645
[36]Zhang X R,Wang L C,Cao Y,et al.Chem.Commnun.,2005,32:4104-4106
[37]XIE Xian-Mei(謝鮮梅),LIU Jie-Xiang(劉潔翔),SONG Jian-Ling(宋建玲),et al.Chin.J.Catal.(Cuihua Xuebao),2003,24(08):569-573
[38]Stoilova D,Koleva V,Vassileva V.Spectrochim.Acta:Part A,2002,58(9):2051-2059
[39]FANG De-Ren(房德仁),LIU Zhong-Min(劉忠民),XU Xiu-Feng(徐瑞峰),et al.J.Fuel Chem.Technol.(Ranliao Huaxue Xuebao),2006,34(1):96-99
[40]CEN Ya-Qing(岑亞青),LI Xiao-Nian(李小年),LIU Hua-Zhang(劉化章).Chin.J.Catal.(CuihuaXuebao),2006,27(3):210-216
[41]Choi Y,Futagami K,Fujitani T.Appl.Catal.A,2001,208(1/2):163-167
[42]Fujitani T,Nakamura.Appl.Catal.A,2000,191(1/2):111-129
[43]LIU Yuan(劉 源),ZHONG Bing(鐘 炳),WANG Qin(王 琴),et al.Chin.J.Catal.(Cuihua Xuebao),1995,16(6):442-446
[44]Reddy B J,Frost R L,Locke A.Transition Met.Chem.,2008,33:331-339
Influence of Microwave Irradiation on Phasetransition in Preparation of Precursor of CuO/ZnO/Al2O3Catalysts for Slurry Methanol Synthesis
FAN HuiLI Zhong*ZHENG Hua-Yan LIU Yan YAN Shao-Wei
(Key Laboratory of Coal Science and Technology,Ministry of Education and Shanxi Province,Taiyuan University of Technology,Taiyuan 030024,China)
The microwave irradiation was used in the aging process of CuO/ZnO/Al2O3catalyst precursor.XRD,FTIR,TG,H2-TPR,HR-TEM and XPS were used to examine the microstructure of precursor and the CuO/ZnO/Al2O3catalyst and investigate the effect of the microvave irradiation on precursor phase transition.The results show that microwave irradiation during the aging process promots the substitution of Zn2+in the Zn5(CO3)2(OH)6compound by Cu2+.During aging process under microwave irradiation,the substitution of Zn2+in Zn5(CO3)2(OH)6compound by Cu2+firstly occurs and completes within 1 h.The Cu2+in Cu2CO3(OH)6compound is constantly substituted by Zn2+with the increasing of aging time,meanwhile,the crystallization degree of(Cu,Zn)5(CO3)2(OH)6becomes higher.Compared with the precursor aged for 1 h without microwave irradiation,the precursor aged for 1 h under microwave irradiation possesses higher content of(Cu,Zn)5(CO3)2(OH)6,the calcined catalysts show strong interaction between CuO and ZnO,well dispersed CuO crystal particles,and high surface CuO content,the methanol space-time yield (STY)and deactivation rate of the catalyst were 318.9 g·kg-1·h-1and 0.11%·d-1,respectively.
microwave irradiation;methanol;slurry reactor;CuO/ZnO/Al2O3catalyst
O614.121;O614.24+
A
1001-4861(2011)03-0509-10
2010-10-25。收修改稿日期:2010-12-03。
國家重點基礎研究發展計劃(973)(No.2005CB221204)資助項目。
*通訊聯系人。E-mail:lizhong@tyut.edu.cn,Tel/Fax:+86-0351-6018526
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50

- 無機化學學報的其它文章
- Synthesis,Crystal Structure and Cytotoxicity of Palladium(Ⅱ)Complexes with N-(4-methylbenzoyl)-L-valine Dianion and Aromatic Diimine
- Synthesis,Crystal Structure of Uranium-Potassium Heteronuclear Coordination Polymer
- Synthesis,Crystal Structure and Antibacterial Activity of Magnesium(Ⅱ)Complex with N-Benzenesulphonyl-L-phenylalanine and 1,10-Phenanthroline
- 鹽湖鹵水萃取提鋰及其機理研究
- 氧化鈦催化羥基磷灰石分解制備可降解磷酸鈣陶瓷
- 菱鎂礦風化石與葉臘石合成堇青石的結構表征