HPLC法測定茜草雙酯固體分散體中的有關物質
2011-08-06 02:57:20肖若蕾
中國醫藥指南 2011年33期
陳 莉 肖若蕾
(咸寧學院藥學院,湖北 咸寧 437100)
茜草雙酯為促進白細胞增生藥,是茜草酸的合成類似物,對各種原因尤其是腫瘤化療所造成的白細胞減少癥有顯著的療效[1]。但茜草雙酯在水中幾乎不溶,且化學性質不穩定。而以PVP為載體制備的茜草雙酯固體分散體能有效的提高茜草雙酯的溶出速率[2],是較理想的茜草雙酯制劑。本文利用HPLC法具有方便簡捷,分離效果好,結果準確,重現性好的優勢,探討了茜草雙酯固體分散體中有關物質的測定方法,以期更好地控制藥品質量。
1 實驗方法
1.1 儀器與試劑
N3000高效液相色譜系統(北京創新通恒),C18色譜柱(250mm×4.6mm,5μm),Sartoriusag BP211D電子天平(北京賽多利斯)。
茜草雙酯固體分散體(自制,每克茜草雙酯固體分散體含0.1g茜草雙酯);茜草雙酯標準品(紫外含量測定用,中國藥品生物制品檢定所,100418-200401);PVPK30(上海化學試劑公司,040914);乙腈為色譜純,其他為分析純,重蒸水自制。
1.2 色譜條件
色譜柱:C18色譜柱(250mm×4.6mm,5μm);流動相為乙腈∶水=70∶30;流速為1.0mL/min;紫外檢測波長為264nm;柱溫:室溫;進樣量為20μL。理論塔板數為8351,重復性試驗符合要求。
1.3 方法的專屬性考察
將茜草雙酯溶液進行強堿、強酸、強氧化、光照等破壞性試驗,考察擬訂的色譜條件下各中間體和降解產物分離情況。
1.3.1 強酸破壞試驗
稱取茜草雙酯對照品約10mg,用20mL流動相溶液溶解,置100mL容量瓶中,加入2.0mL的鹽酸溶液(1.0mol/L),振搖,于80℃水浴中加熱30min,取出,冷卻至室溫,用氫氧化鈉溶液(0.5mol/L)調pH=5,再用流動相溶液稀釋至刻度,搖勻。
1.3.2 強堿破壞試驗
稱取茜草雙酯對照品約10mg,用20mL流動相溶液溶解,置100mL容量瓶中,加入2.0mL的氫氧化鈉溶液(1.0mol/L),振搖,于80℃水浴中加熱30min,取出,冷卻至室溫,用的鹽酸溶液(0.5mol/L)調pH=5后,再用流動相溶液稀釋至刻度,搖勻。
1.3.3 氧化破壞試驗
稱取茜草雙酯對照品約10mg,用20mL流動相溶液溶解,置100mL容量瓶中,加入30%的過氧化氫溶液2.0mL,振搖,于80℃水浴中加熱30min,取出,冷卻至室溫,再用流動相溶液稀釋至刻度,搖勻。
1.3.4 強光照射破壞試驗
稱取茜草雙酯對照品約10mg,用適量流動相溶液溶解,置100mL容量瓶中,置陽光下照射3h,用流動相溶液稀釋至刻度,搖勻。
分別取上述方法破壞后的茜草雙酯溶液適量,微孔濾膜過濾,按1.2項下色譜條件進樣,觀察主峰與各雜質峰的分離情況,結果如圖1。
由破壞性實驗的高效液相色譜圖可知,在擬訂的色譜條件下,有關物質和降解產物的色譜峰出峰在主峰前,有關物質及輔料色譜峰與主成分茜草雙酯峰可以有效分離。據此,將有關物質檢查的記錄時間擬訂為主峰保留時間的2倍。實驗結果表明:堿性條件下茜草雙酯尤其不穩定,茜草雙酯只有極少量存在,破壞產物極性較強,在本色譜條件下,基本不保留;茜草雙酯在其他破壞性實驗條件下有關物質和降解產物也明顯增加,因反應機理較復雜,未能確定其主要降解產物的化學結構。
1.4 線性關系試驗
精密稱取茜草雙酯標準品適量,用流動相溶解配制成100.0μg/mL的對照品貯備液。分別移取不同體積的對照品貯備液于10mL棕色容量瓶中定容,搖勻。按1.2項下色譜條件進樣,并測定。記錄色譜流出曲線,以峰面積(A)對濃度C進行線性回歸,茜草雙酯在濃度10.0~100.0μg/mL范圍內呈良好線性關系,回歸方程為A=2.76×104C+8.03×103,r=0.9997。
1.5 最低檢測限
按信噪比為3(S/N=3)作為最低檢測限,以濃度為10ng/mL,進樣20μL時,主產物峰信號約為基線噪音的3倍,即最低檢測限為0.2ng。
1.6 重復性試驗
精密稱取同批茜草雙酯固體分散體,共6份(約含茜草雙酯5mg),置50mL容量瓶中,用流動相溶液稀釋至刻度,搖勻,再從中移取5mL至10mL棕色容量瓶中定容,搖勻。按1.2項下色譜條件測定,記錄色譜圖。結果:茜草雙酯平均含量為97.2%,RSD=1.02%(n=6)。

圖1 茜草雙酯及其破壞性實驗的高效液相色譜圖
1.7 溶液的穩定性
精密稱取茜草雙酯固體分散體適量(約含茜草雙酯5mg),溶于流動相溶液,置50mL容量瓶中,用流動相溶液稀釋至刻度,搖勻,將所得樣品溶液在室溫且避光條件下放置0,1,2,3,4h后分別取樣測定,得主峰峰面積的RSD為0.29%,表明茜草雙酯溶液在4h內穩定。
2 茜草雙酯固體分散體中有關物質的測定
2.1 茜草雙酯固體分散體的制備
茜草雙酯-PVP固體分散劑,將茜草雙酯與PVP以1∶5的比例溶于適量的無水乙醇,經減壓蒸發除去乙醇,在干燥器內平衡數日,粉碎過八十目篩,得固體分散體樣品[2],備用。
2.2 試樣溶液的配制
精密稱取不同次制備的茜草雙酯固體分散體各一份(約含茜草雙酯5mg),置50mL容量瓶中,用流動相溶液稀釋至刻度,搖勻。按1.2項下色譜條件測定,記錄色譜圖至主成分保留時間的2倍,供試品如顯雜質峰,按峰面積歸一化法計算雜質含量。結果如表1。
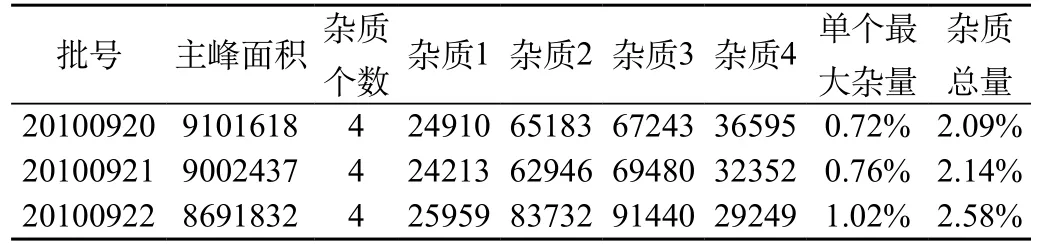
表1 茜草雙酯固體分散體中有關物質測定結果
3 討 論
有關物質的檢查是保證藥品安全有效的重要指標之一,由于茜草雙酯化學性質不穩定,易發生氧化和水解等化學反應,其原料藥及制劑的有關物質檢查更不容忽視,因此,必須選擇專屬性強、靈敏度高、重復性好的方法。茜草雙酯原料藥的有關物質檢測HPLC方法已經建立。本研究建立的HPLC方法測定茜草雙酯固體分散體中有關物質的方法表明主峰與各雜質峰均能達到較好分離,具有靈敏度高的優點,可用于茜草雙酯固體分散體中有關物質測定。
實驗過程中比較了不同的溶劑,包括:乙醇-水,無水乙醇,甲醇,乙腈及本實驗所用的流動相,由于茜草雙酯化學性質不穩定,易氧化和水解及不易溶于水的特點,同時可以減少溶劑峰對圖譜效果的影響,便于觀察與區分雜質峰,最終確定選擇流動相作溶劑,并在實驗過程應注意避光處理,此時樣品可較好溶解,且在4h內穩定。
本實驗比較了各種流動相對分離效果的影響∶甲醇-水-四氫呋喃=70∶30∶5,甲醇-水-四氫呋喃=70∶20∶5,甲醇-水=80∶20,甲醇:水=70:20,乙腈:水:四氫呋喃=70∶30∶5,乙腈∶水=70∶30,得出當流動相為乙腈∶水=70∶30時,茜草雙酯及有關物質的色譜峰峰型、分離度均較好,拖尾因子小,出峰時間合適。
[1] 王升啟,馬立人.茜草屬藥用植物的化學成分及生物活性[J].軍事醫學科學院院刊,1991,14(4):254-259.
[2] 肖若蕾,陳莉.茜草雙酯固體分散體的制備及體外溶出實驗[J].解放軍藥學學報,2009,25(5):457-459.
