托拉塞米片溶出度的方法學研究*
2011-06-21 03:04:58趙洪霞
天津藥學 2011年4期
趙洪霞
(天津安捷倫藥業有限公司,天津 300410)
托拉塞米是新一代高效髓袢利尿劑。多年臨床應用證實,托拉塞米適應證廣,利尿作用迅速強大且持久,不良反應發生率低,是臨床上值得推廣的一類高效利尿劑。參照托拉塞米標準(試行)YBH00682004,依照《中國藥典》溶出度測定法[1],為了操作簡便,進行了適當改進,采用紫外可見分光光度法(UV)測定本品的溶出度。
1 試驗材料
1.1儀器 Waters 2487紫外分光光度計,ZRS-8G智能溶出試驗儀(天津大學無線電廠),日本島津LC-10A系列高效液相色譜儀,XS204型電子天平(瑞士梅特勒-托利多公司)。
1.2試藥 托拉塞米對照品(本公司自制,批號100206,純度:99.5%),托拉塞米片(本公司自制,批號100301、100302、100303)。鹽酸(天津市申泰科密歐化學試劑有限公司 分析純),磷酸二氫鉀(天津市醫藥公司 分析純),冰乙酸(天津市贏達稀貴化學試劑廠 分析純)。
2 試驗和結果
2.1溶出介質的選擇 取托拉塞米片6片,照溶出度測定法(《中國藥典》2010年版二部附錄Ⅹ C第二法)分別以0.1 mol/L鹽酸溶液 、水、磷酸鹽緩沖液(pH=6.8)各900 ml為溶出介質,轉速為50 r/min,分別在5、10、15、20、30 min取溶液適量,濾過,取續濾液照紫外-可見分光光度法(《中國藥典》2010年版附錄二部Ⅳ A),在285 nm的波長處測定吸光度;另精密稱取托拉塞米對照品適量,加冰乙酸2 ml,振搖使溶解,再加溶出介質制成每1 ml中約含10 μg的溶液,同法測定,計算每片的溶出量,繪制不同溶出介質的溶出曲線,見圖1。試驗結果表明:溶出介質為0.1 mol/L鹽酸溶液、水、磷酸鹽緩沖液(pH=6.8)時,托拉塞米片均可達到最大溶出,以0.1 mol/L鹽酸溶液為溶出介質溶出曲線較平滑,且參照托拉塞米片進口標準H20030655、H20030656選擇標準中使用的鹽酸作為溶出介質。
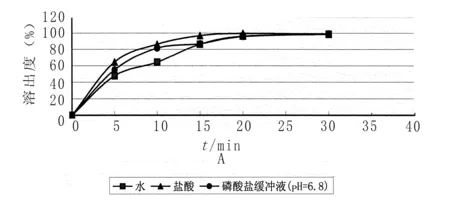
圖1 不同溶出介質溶出曲線圖
2.2攪拌方式的選擇 取托拉塞米片樣品6片,照溶出度測定法(《中國藥典》2005二部附錄Ⅹ C第一法)以0.1 mol/L鹽酸溶液900 ml為溶出介質,轉速為100 r/min,分別在5、10、15、20、30 min取溶液適量,濾過,取續濾液照紫外-可見分光光度法(《中國藥典》2010年版附錄二部Ⅳ A),在285 nm的波長處測定吸光度;另取托拉塞米對照品適量,加冰乙酸2 ml,振搖使溶解,再加溶出介質稀釋成每1 ml中約含10 μg的溶液,同法測定,計算每片溶出量,繪制溶出曲線,見圖2。試驗結果表明:采用以上兩種攪拌方式,溶出均達到最大值,參照托拉塞米片進口標準H20030655、H20030656,故采用標準中的攪拌方式即槳法。
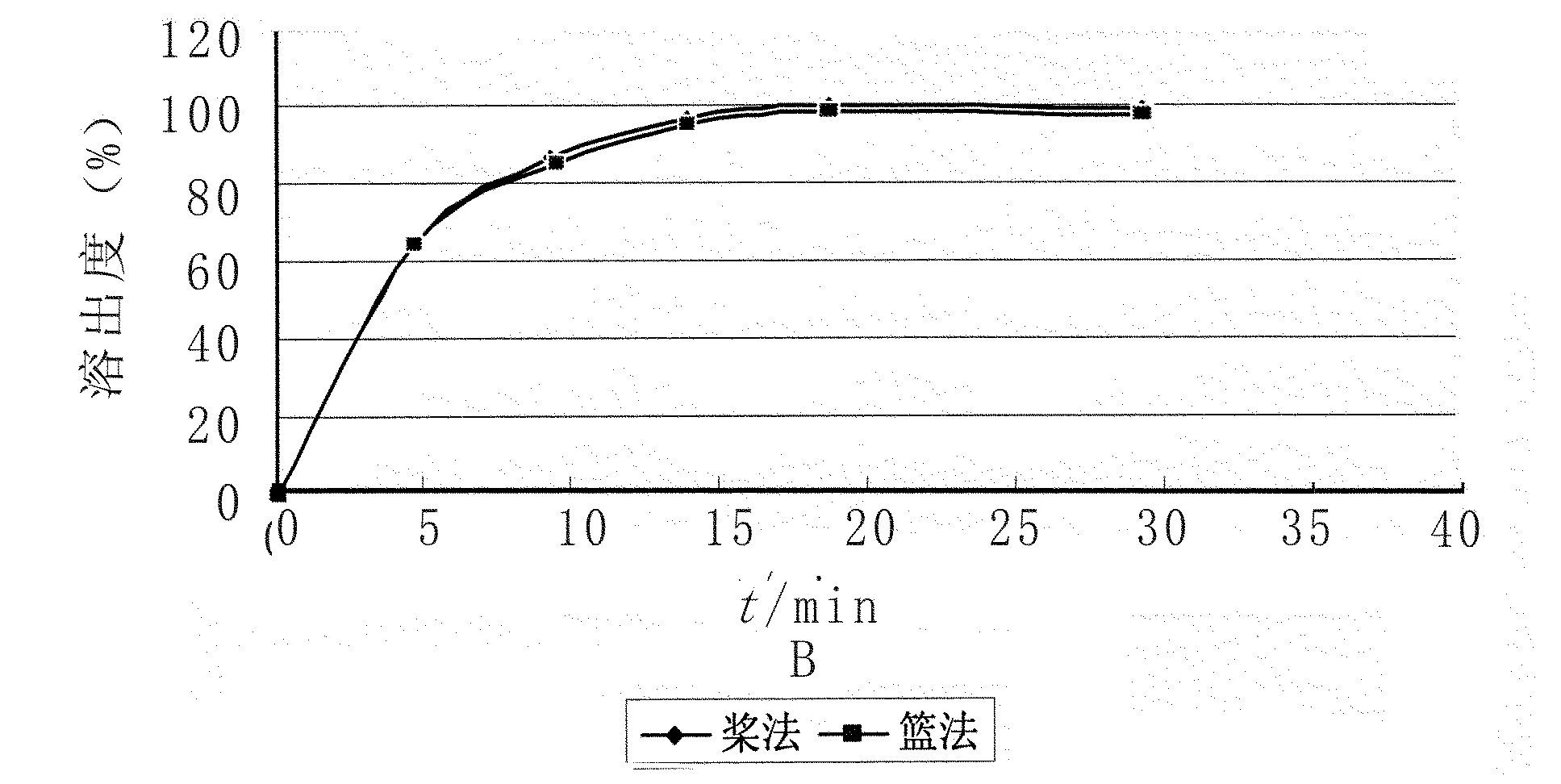
圖2 溶出度槳法、籃法溶出曲線圖
2.3藥物在溶出介質中的線性試驗 取托拉塞米原料20 mg, 精密稱定,置100 ml量瓶中, 加冰乙酸2 ml,振搖使溶解,再加0.1 mol/L鹽酸溶液稀釋至刻度,搖勻,濾過,精密量取續濾液5 ml置50 ml量瓶中,加0.1 mol/L鹽酸溶液稀釋至刻度,搖勻;精密量取上述溶液2、4、6、8、10 ml分別置于10 ml量瓶中,加0.1 mol/L鹽酸溶液稀釋至刻度,搖勻。照紫外-可見分光光度法(《中國藥典》2010年版附錄二部Ⅳ A),在285 nm的波長處分別測定吸光度,進行線性回歸。結果表明:在4~20 μg/ml的濃度范圍內,托拉塞米的濃度與吸光度呈良好的線性關系,線性方程為Y=0.032X+0.023(r=0.999 5)。
2.4回收率試驗 取托拉塞米原料適量,精密稱定,按處方配比加入適量輔料,用溶出介質制成8、10、12 μg/ ml的梯度各3份,搖勻。照紫外-可見分光光度法(《中國藥典》2010年版附錄二部Ⅳ A),在285 nm的波長處分別測定吸光度,記錄數據,計算托拉塞米的回收率,結果托拉塞米在該溶出介質中的平均回收率為99.2%,RSD為0.49%。見表1。
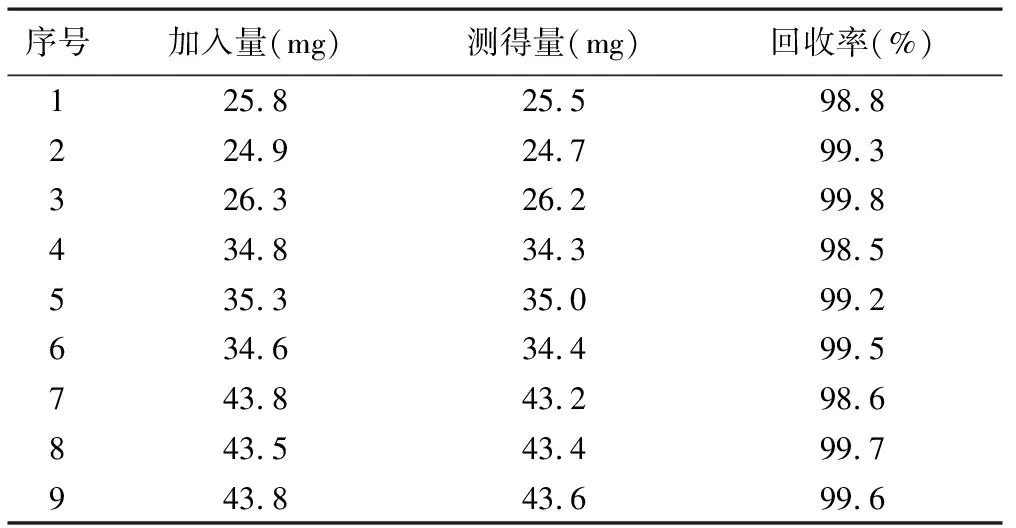
表1 托拉塞米片回收率試驗的測定結果
2.5托拉塞米片溶出液的穩定性試驗 取溶出度測定的同一溶液,于不同時間測定吸收度,RSD為0.6%。結果表明,樣品在溶出介質中8 h內穩定。
2.6均一性試驗 取100301批樣品6片,照溶出度測定法(《中國藥典》2005二部附錄Ⅹ C第二法)以0.1 mol/L鹽酸溶液900 ml為溶出介質,轉速為50 r/min,分別在5、10、15、20、30 min取溶液適量,濾過,取續濾液照紫外-可見分光光度法(《中國藥典》2010年版附錄二部Ⅳ A),在285 nm的波長處分別測定吸光度,另取托拉塞米對照品適量,精密稱定,加冰乙酸2 ml,振搖使溶解,再加溶出介質稀釋成每1 ml中約含10 μg的溶液,同法測定,計算每片的溶出量,見圖3。從圖3可以看出,本方法測定的溶出度均一性良好。
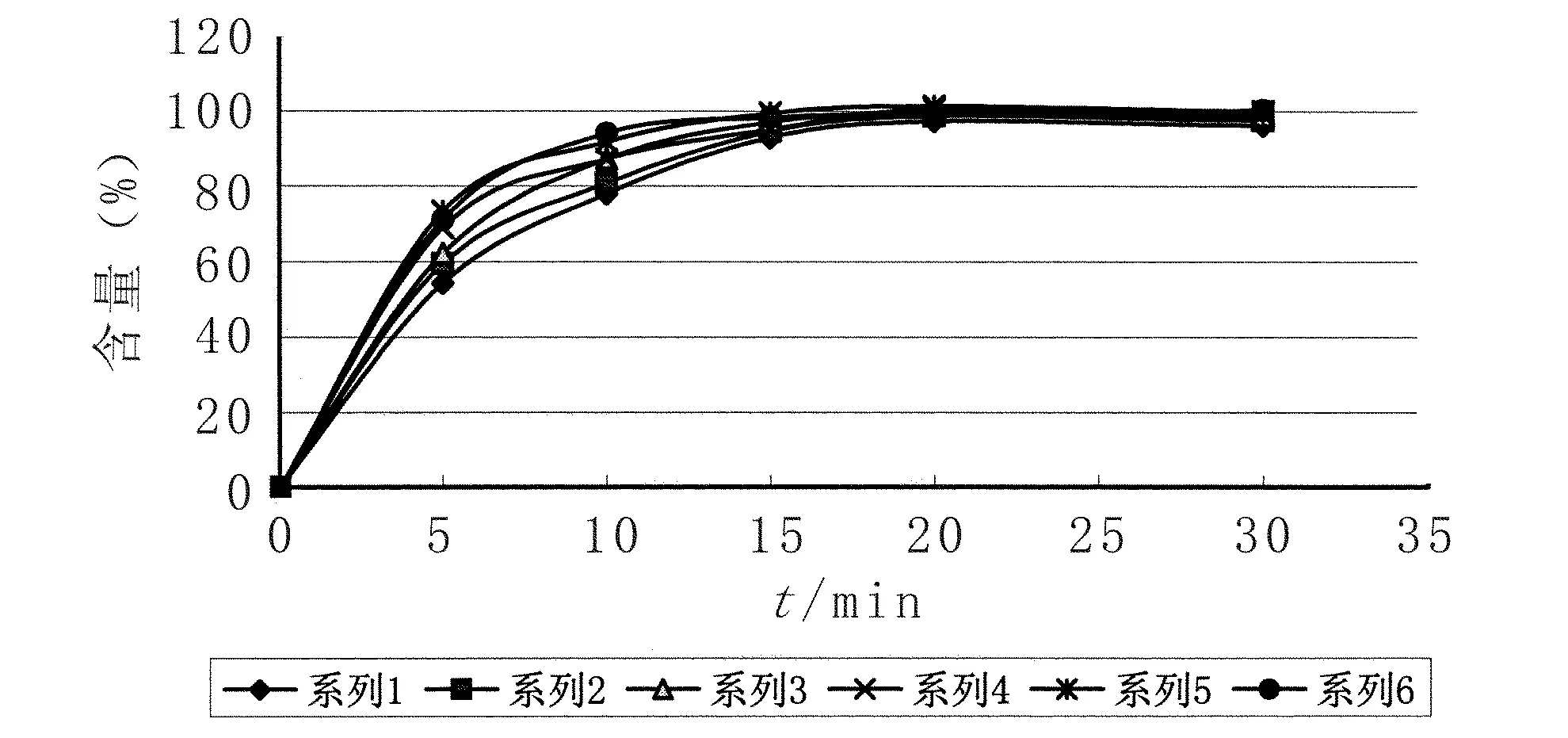
圖3 托拉塞米片100301批樣品溶出曲線
2.7樣品溶出曲線的測定 試驗結果表明,3批樣品溶出曲線差異較小,符合規定。見圖4。
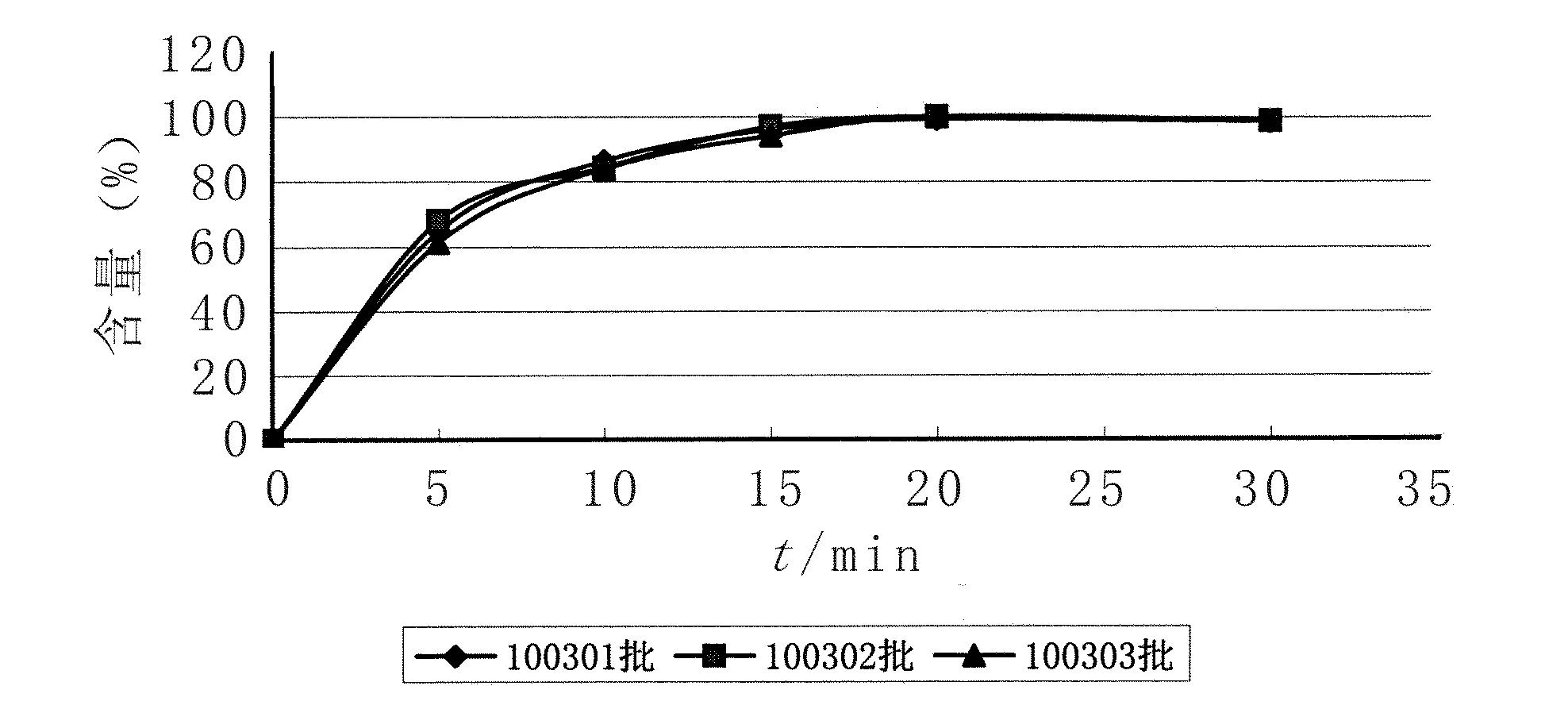
圖4 3批樣品溶出曲線
2.8兩種方法測定溶出度的結果比較 取本品,照溶出度測定法 (《中國藥典》2010年版二部附錄Ⅹ C第二法),以0.1 mol/L鹽酸溶液900 ml為溶出介質,轉速為50 r/min,依法操作,20 min時,取溶液適量,濾過,取續濾液,照紫外-可見分光光度法(《中國藥典》2010年版附錄二部Ⅳ A),在285 nm的波長處測定吸光度;另取托拉塞米對照品適量,精密稱定,加冰乙酸2 ml,振搖使溶解,再加溶出介質稀釋成每1 ml中約含10 μg的溶液,同法測定,計算每片溶出量,限度為標示量的80%,應符合規定。依照進口標準H20030655.H20030656進行高效液相色譜法測定。結果表明:采用紫外可見分光光度法和依照進口標準H20030655.H20030656高效液相色譜法測定的本品溶出度結果無明顯差異。見表2。
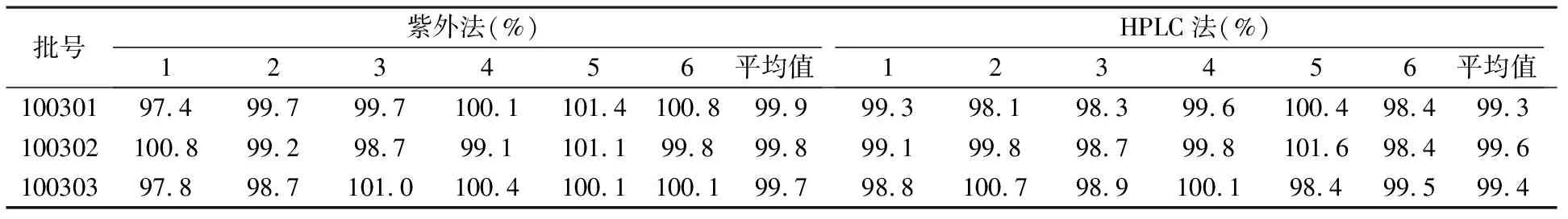
表2 紫外法和HPLC法測定結果比較
3 討論
3.1測定波長的選擇 依據托拉塞米標準(試行)YBH00682004,取托拉塞米對照品適量,加適量冰乙酸使溶解,再加0.1 mol/L鹽酸溶液制成每1 ml中約含10 μg 的溶液,照紫外-可分光光度法(《中國藥典》2010年版二部附錄Ⅳ A)測定,該溶液在285、210 nm的波長處有最大吸收,因托拉塞米在285 nm波長處波峰較為平緩,故選擇在285 nm的波長處測定。
3.2冰酸的干擾試驗 取2 ml冰乙酸,置100 ml量瓶中,用0.1 mol/L鹽酸溶液稀釋至刻度,搖勻;再精密量取5 ml置100 ml量瓶中,用0.1 mol/L鹽酸溶液稀釋至刻度,搖勻。取此溶液在200~400 nm的波長范圍內進行紫外掃描。結果表明,冰乙酸在285 nm的波長處沒有吸收,冰乙酸對托拉塞米吸光度測定干擾為陰性。
3.3濾膜的吸附試驗 取托拉塞米約20 mg,精密稱定,置100 ml量瓶中,加冰乙酸2 ml,振搖使溶解,再加0.1 mol/L鹽酸溶液稀釋至刻度,搖勻;精密量取5 ml置100 ml量瓶中,加0.1 mol/L鹽酸溶液稀釋至刻度,搖勻;在285 nm的波長處分別測定其吸光度,照上述方法共稱取6份,進行測定,分別用水膜濾過,同法測定,結果顯示:托拉塞米過水膜吸光度與未過膜吸光度的比值為0.995 7,RSD為0.52%。試驗結果表明,水膜對托拉塞米無明顯吸附,可忽略不計。
3.4輔料的干擾性試驗 按處方配比,取約相當于1片重量的輔料壓片,共六片,取上述空白片,照溶出度測定法(《中國藥典》2010二部附錄XC第二法)以0.1 mol/L鹽酸溶液900 ml為溶出介質,轉速為50 r/min,依法操作,在20 min時,取溶液適量,濾過,取續濾液作為供試品溶液;另取托拉塞米對照品適量,精密稱定,加冰乙酸2 ml,振搖使溶解,再加溶出介質稀釋成每1 ml中約含10 μg的溶液,作為對照品溶液。取上述溶液,照紫外-可見分光光度法(《中國藥典》2010年版附錄二部Ⅳ A),在285 nm的波長處分別測定吸光度,結果顯示輔料干擾率為0.85%小于2.0%。以上試驗結果表明:輔料對溶出結果測定影響較小,可忽略不計。
本試驗采用紫外-可見分光光度法測定托拉塞米片溶出度,并對所建立的方法進行了方法學的驗證,線性范圍為4~20 μg/ml(r=0.999 5),回收率為99.5%,RSD為0.42%;所得結果與高效液相色譜法進行溶出度測定的結果比較沒有明顯差異,是一種快速,簡便,準確的分析方法,可作為托拉塞米片溶出度測定的檢測方法。
1 中國藥典.二部.2010:附錄:85
