SUMO基因的內在原核啟動子活性及其在大腸桿菌蛋白表達系統中的應用
2011-02-10 01:20:30亓燕紅鄒竹榮鄒華英范云六張春義
生物工程學報 2011年6期
關鍵詞:融合
亓燕紅,鄒竹榮,鄒華英,范云六,張春義
1 山東農業大學農學院,泰安 271000
2 云南師范大學生命科學學院,昆明 650092
3 云南省農業科學院 院辦檔案科,昆明 650231
4 中國農業科學院生物技術研究所,北京 100081
在真核生物泛素家族成員中,SUMO (Small ubiquitin-related modifier,小泛素相關修飾物) 具有多種重要的生物學功能。SUMO化修飾介導靶蛋白分子定位和功能調節,在轉錄調控、核質轉運、信號轉導、DNA修復和細胞周期調控等方面均發揮著重要作用[1-2]。
近年來,SUMO作為N端融合標簽在大腸桿菌表達系統中得到越來越多的應用[3-8]。SUMO標簽不僅能有效提高目標蛋白的表達水平和可溶性[5],而且整體作為一個空間結構上的識別位點,被其特異性蛋白酶 Ulp1從融合表達蛋白上定位 (SUMO末端二甘氨酸,-GG基序的后隨肽鍵上) 切除[3,9],不存在任何非特異性酶切,目標蛋白上可以不留任何殘基,這對生產具有天然氨基酸序列的重組蛋白極其適用。另外,Ulp1不僅酶切效率高,而且在較廣范圍的pH和溫度條件下都具有活性[3],這相比其他切除標簽、依賴識別序列的工具蛋白酶 (如Factor Xa、煙草蝕刻病毒蛋白酶 TEV、腸激酶 EK) 都具有顯著的綜合優勢。類似的泛素 (Ubiquitin) 融合系統雖然具備 SUMO融合系統的諸多優點[3,10],但切除泛素標簽的去泛素化酶DUBs不穩定,難以生產和廉價制備[3];而且大腸桿菌中存在一種內源性泛素蛋白酶 ElaD,特異性切割泛素融合蛋白,使得泛素標簽作用失去意義[11]。因此,SUMO融合技術已成為目前大腸桿菌重組蛋白表達和純化的重要手段。
盡管如此,SUMO融合系統在載體構建效率和蛋白可溶性等方面仍有待改進。在本實驗中,我們發現釀酒酵母SUMO基因Smt3(Sm) 具有組成型原核啟動子活性。利用此特性,結合引入Sm 3′末端StuⅠ位點以及His標簽和超酸增溶標簽,構建了基于 Sm’-LacZα融合基因的系列通用克隆表達載體。通過多個基因克隆和表達實驗,結果表明在改良的SUMO融合系統中實現了基因表達載體的快速構建和蛋白高可溶性表達,一同建立的共表達載體系統還可以用作研究細胞內蛋白間的相互作用。
1 材料與方法
1.1 材料
大腸桿菌菌株 DH5α、BL21(DE3) 以及pUC19、pET32a(+)、pACYC184和pEGFP-N1等質粒為本實驗室保存,本工作中所建載體以外的其他載體均為本實驗室先前構建和保存。限制性內切酶、pfu DNA聚合酶、T4 DNA連接酶和 DNA marker購自MBI Fermentas公司;DNA快速純化回收試劑盒和蛋白分子量marker購自北京鼎國生物技術有限公司;氨芐青霉素 (Amp)、氯霉素 (Cm)、異丙基-β-D-硫代半乳糖苷 (IPTG) 為Genview公司產品;其他生化試劑均為國產分析純。本工作中所有引物 (表1) 的合成以及DNA序列分析均由上海生工生物工程技術服務有限公司完成。

表1 本研究中所用引物Table 1 Primers used in this study
1.2 方法
1.2.1 載體的構建
所用基因操作方法均參照分子克隆實驗手冊[12]。載體構建如結果中詳述,并通過DNA測序驗證。
1.2.2 原核啟動子預測分析
在NCBI網站上查詢并提取各物種SUMO基因的 mRNA序列,分別對其基因編碼區序列 (CDS)通過軟莓網站 (www.softberry.com) 提供的原核啟動子預測程序 BPROM 和設定的參數 (LDF≥0.2指含有啟動子) 進行分析,得到預測原核啟動子的強度 (LDF值)、?10和?35元件等信息,并通過EXCEL軟件整理分析。
1.2.3 蛋白表達及SDS-PAGE分析
將表達載體轉化至大腸桿菌BL21(DE3) 中,挑取單菌落37 ℃培養過夜。然后按照4%量接種到含相應抗生素濃度的LB液體培養基中 (對pET載體單獨表達使用100 mg/L Amp,對pAY載體單獨表達使用34 mg/L Cm;對pET和pAY載體共表達使用100 mg/L Amp和34 mg/L Cm),37 ℃培養至OD600達0.6,加入IPTG至終濃度1 mmol/L,37 ℃誘導表達4 h。收集10 mL的誘導表達細菌培養液,離心后加入3 mL PBS緩沖液懸浮,并用超聲波裂解。取細胞裂解物20 μL,15 000 r/min、4 ℃離心5 min,分為上清 (S) 和沉淀 (P),用12% SDS-PAGE進行電泳檢測,并用凝膠成像系統生成圖形文件。
1.2.4 蛋白體外酶切反應
將SUMO融合蛋白的pET載體表達細菌與酵母Ulp1蛋白酶的載體pET(Ulp1) 表達細菌分別按上述方法制備得到裂解物上清。兩者上清一般按體積比20∶1混勻,放置 25 ℃反應 3 h,然后用 12% SDS-PAGE分析反應情況。
2 結果
2.1 酵母SMUO基因Smt3具有組成型原核啟動子活性
利用引物 SmNdFw/SmStRv擴增釀酒酵母SUMO基因 Smt3(Sm) 的 DNA片段平末端克隆到pUC19的HincⅡ(GTCGAC) 位點,轉化大腸桿菌DH5α,發現在X-gal+IPTG的LB平板上長出來的菌落幾乎全都為藍色。經PCR鑒定Sm的重組克隆,命名為pUC(Sm-LacZα)。

圖1 質粒pUC19(Sm-LacZα) 中SUMO基因Smt3與LacZα融合的序列分析Fig. 1 Sequence analysis of the fusion gene of Smt3 and LacZα in plasmid pUC19 (Sm-LacZα).
通過測序分析,發現Sm插入 (圖1,兩豎箭頭之間) 打斷了pUC19上LacZα的翻譯閱讀框,即由核糖體結合位點 RBS(lac) [AGGA]介導的翻譯 (圖1)。但是,Sm的反向引物SmStRv在其3′末端 (即 -GG基序密碼子處) 引入 StuⅠ位點 (AGGCCT)產生了一個經典的RBS,即RBS(sm) [GGAGG],同時該引物 5′端附加保護堿基對應的互補堿基 (-AT 3′) 與克隆位點HincⅡ的平末端 (5′ GAC-) 融合產生一個ATG密碼子,與RBS(Sm) 的間隙長為7 bp,這些剛好為其下游LacZα表達提供了有效的翻譯元件 (圖 1),從而導致藍色菌落形成。不過,這種翻譯模板除了由Lac啟動子 (Plac) (圖1) 在IPTG誘導下產生的Sm-LacZα轉錄產物,會不會在Sm上存在其他啟動子產生新的轉錄產物并由 RBS(sm) 介導翻譯LacZα呢?
通過對 Sm(306 bp) 進行啟動子預測分析,發現 Sm存在依賴σ70的原核啟動子 Psm,包含?35(Psm)、?10(Psm) 元件,分別為 TTAAGA、TATTAGAAT (其后面6 bp為核心序列) (圖1),可能引導下游LacZα表達。由于受固有Lac啟動子的轉錄干擾,這種預測需要進一步證實。
隨后以 pUC(Sm-LacZα) 為模板,利用引物SmNdFw和 LZXhRv,擴增出 Sm-LacZα片段,經NdeⅠ和 XhoⅠ酶切,與同樣酶切的 pET32a(+) 連接,轉化大腸桿菌DH5α,發現在X-gal+IPTG的LB平板上也能長出藍色菌落,而且經鑒定全都為陽性克隆,取名pET(Sm-LacZα)。由于pET32a(+) 使用依賴噬菌體 T7 RNA聚合酶的 T7啟動子 (后隨LacO序列),DH5α和載體自身不表達T7 RNA聚合酶,因而即便存在 IPTG也不會由 T7啟動子轉錄Sm-LacZα,那么這個導致藍色菌落形成的LacZα表達唯獨可能由Sm啟動子Psm的作用所致。
另外,以pEGFP-N1為模板,利用引物GFPfw和GFPrv擴增GFP (綠色熒光蛋白基因) 編碼區片段,分別平端插入到 pET(Sm-LacZα) 的 StuⅠ和Ecl136Ⅱ位點 (圖1),轉化大腸桿菌DH5α,通過藍白斑篩選和菌落 PCR 鑒定正確陽性克隆pET(Sm-GFP) stu和pET(Sm-GFP) ecl。GFP片段在兩個位點的插入都導致其后面的LacZα不表達,形成白色菌落。分別挑取 2種白菌落劃線在普通 LB平板上培養,發現pET(Sm-GFP) ecl的菌落逐漸變成綠色,而pET(Sm-GFP) stu的菌落顏色則沒什么變化。通過序列分析,GFP在兩個位點的插入都與其5′端Sm形成一個正確閱讀框的融合基因,而且都有可能由Sm啟動子引導GFP轉錄物產生。但是,在StuⅠ位點插入導致RBS (sm) (圖1) 與GFP起始密碼子ATG緊密相鄰,GFP不能在此處翻譯;而在Ecl136 II位點插入則可以通過RBS (sm) 和其后的ATG翻譯出GFP蛋白并累積,使pET(Sm-GFP) ecl菌落逐漸變成綠色。這表明在誘導物IPTG不存在的情況下,Sm啟動子能夠組成型發揮轉錄活性,疊加在 StuⅠ位點的 RBS(sm) 是其下游基因翻譯的重要功能元件。總之,上述結果表明酵母SMUO基因Smt3具有明顯的組成型原核啟動子活性。
2.2 不同物種SUMO基因的原核啟動子預測分析
SMUO是真核生物中廣泛存在的一類蛋白,釀酒酵母SUMO基因Smt3具有原核啟動子活性,我們推測其他物種的SUMO基因可能具有同樣特性。
通過軟莓程序BPROM對NCBI核酸數據庫中幾乎所有的SUMO基因編碼區 (CDS) 進行啟動子預測分析,發現121個被測對象中約90%都具有原核啟動子活性 (LDF≥0.2),在不同范圍 LDF值的SUMO CDS的比例依次為3% (>5)、4% (4~5)、9% (3~4)、19% (2~3)、38% (1~2) 和17% (0.2~1) (圖2)。進一步將不同SUMO CDS按物種劃分成4類:真菌 (Fungi)、植物 (Plant) (絕大多數為被子植物)、無脊椎動物 (Invertebrate) 和脊椎動物(Vertebrate),然后分類統計它們在不同LDF范圍所占比例。結果顯示,真菌和植物中 85%的 SUMO CDS具有原核啟動子活性,而在脊椎和無脊椎動物中這個比例高達95%。LDF>3的SUMO CDS在各類物種所占比例分別為30% (真菌)、18% (植物)、27% (無脊椎動物) 和2% (脊椎動物),說明除脊椎動物外其他物種的 SUMO CDS有較大比例具有較高的原核啟動子活性,尤其在真菌和無脊椎動物中表現更為突出,LDF>4的比例分別為19%和16% (圖2)。
2.3 基于Sm’-LacZα融合基因的通用克隆表達載體構建及其特性分析

圖2 含某指定范圍預測啟動子LDF值的SUMO基因CDS在同類物種中所占比例Fig. 2 Percentages of SUMO CDSs with indicated range of LDFs of the predicted promoters in the same category of species.
以 pET(Sm-LacZα) 為模板,使用引物對HSNdFw/SmNsRv和LZNsFw/LZXhRv,通過引物重疊延伸法[13]合成新的融合基因片段 Sm’-LacZα。通過NdeⅠ和XhoⅠ酶切,與同樣酶切的pET32a(+) 連接,構建成pET(Sm’-LacZα) (圖3A)。Sm’是由引物HSNdFw使Sm 5′端加上了一個His標簽序列 (H6),有利于表達蛋白純化;同時在 Sm’-LacZα融合處又引入了NcoⅠ和SalⅠ/HincⅡ位點,使得該處多克隆位點 (MCS) 數目更多,便于目的基因 (如TEV) 多種形式克隆和表達。在Sm 3′末端 -GG基序密碼子處,引入了一個StuⅠ位點下劃線代表-GG基序密碼子,代表Ulp1切點;大寫序列 AGGCCT代表 StuⅠ位點,代表平末端切點) (圖1、圖3A),該酶切平末端5′-Sm’ ggAGG-3′能與任何靶蛋白編碼序列 target的 PCR產物 5′-N target-3′ (在target 5′端僅需通過PCR引物添加任何一種堿基N,表1) 連接,即可得到正確閱讀框的Sm’-target融合基因 (即5′-Sm’ ggAGGNtarget-3′),融合表達蛋白剛好在-GG基序 (由ggAGGN 編碼)后被 Ulp1酶切開,不會殘留任何氨基酸在靶蛋白的N端。由于Sm標簽還只能相對有限地提高靶蛋白的可溶性[7-8],我們分別將超酸增溶標簽 Msb (E. coli msyB)、Yd (E. coli yjgD)、Od (E. coli rpoD的N端結構域)[7-8]的PCR產物用NdeⅠ酶切,然后插入到pET(Sm’-LacZα) 的NdeⅠ位點 (圖3A),構建成通用增溶表達載體 pET(MsbSm’-LacZα)、pET(YdSm’-LacZα) 和pET(OdSm’-LacZα),超酸標簽Msb、Yd、Od與Sm’分別形成復合超酸增溶標簽MsbSm’、YdSm’和OdSm’。由于Sm原核啟動子活性,所有這些載體的大腸桿菌 DH5α轉化菌落在X-gal+IPTG平板上都呈現藍色。總之,這些載體組成的改良SUMO融合系統理論上不但通過藍白斑篩選非常便于目的基因PCR產物的快速克隆和蛋白可溶性表達,而且尤其適合含天然氨基酸序列靶蛋白的重組生產。
此外,用引物PayFw和PayRv擴增低拷貝型質粒 pACYC184[14],得到去除四環素抗性基因的大片段,然后用EcoR V酶切,與上面Sm’-LacZα融合基因 PCR片段連接,轉化大腸桿菌 DH5α,通過藍白斑篩選和菌落 PCR鑒定構建了重組質粒pAY(Sm’-LacZα) (圖 3B)。與載體 pET(Sm’-LacZα)一樣,它也可以通過藍白斑篩選用作目的基因片段(如Ulp1) 的快速克隆和表達。由于pAY(Sm’-LacZα)派生載體含pACYC184的復制起始區p15A,與其他類型如pET(Sm’-LacZα) 派生載體 (含 ColEI復制起始區) 相容,兩者可在同一細菌中共表達,可以用于兩個 (或幾個) 表達靶蛋白的相互作用分析。
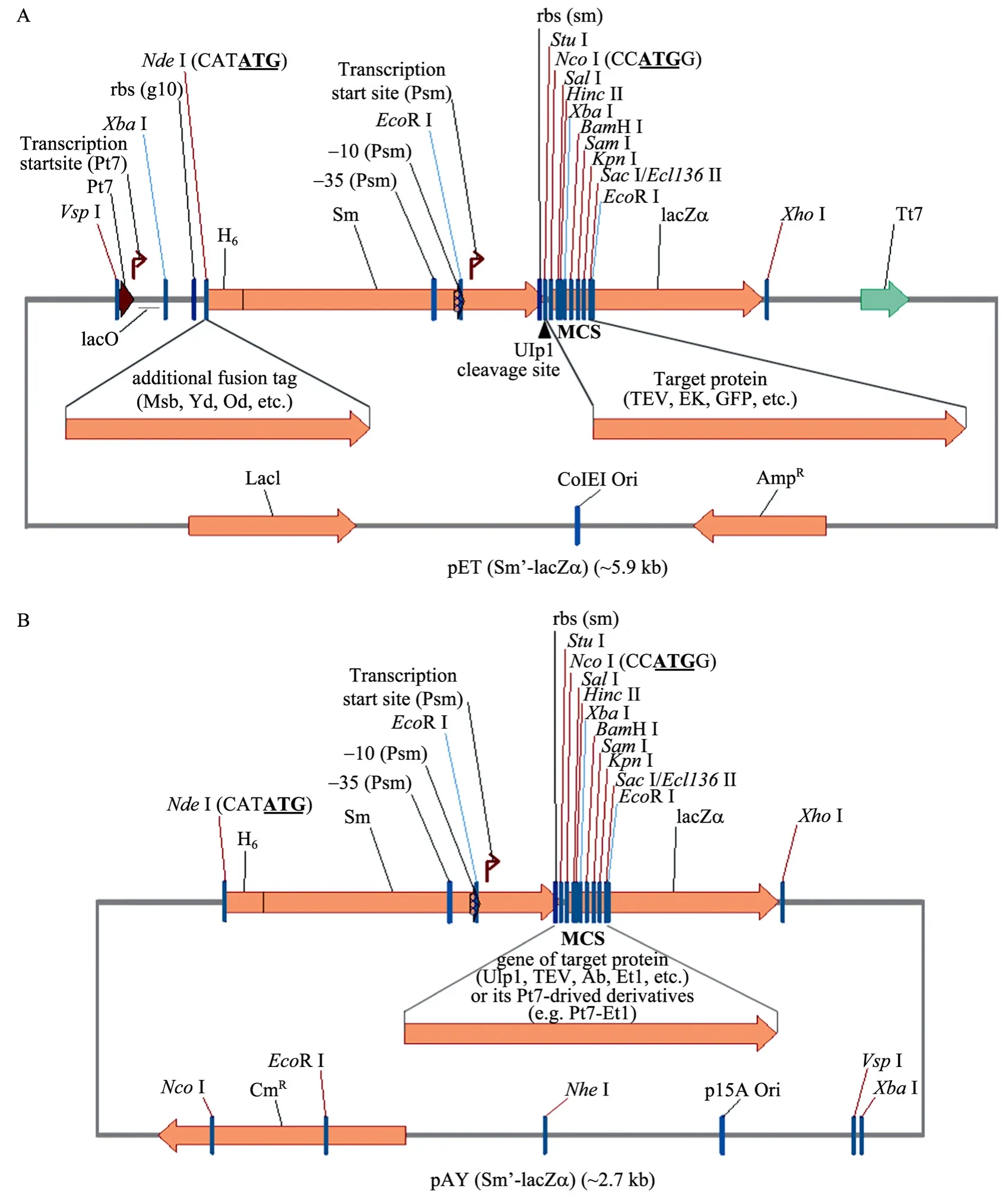
圖3 載體pET(Sm’-LacZα) (A) 和pAY(Sm’-LacZα) (B) 示意圖Fig. 3 Diagrams of vectors pET(Sm’-LacZα) (A) and pAY(Sm’-LacZα) (B).
2.4 基于 pET(Sm’-LacZα) 系列的基因表達載體構建及其蛋白表達分析
根據上述 pET(Sm’-LacZα) 系列載體的特性,分別利用引物對 GfpFw/GfpRv、Et1Fw/Et1Rv、TevFw/TevRv、EkFw/EkRv從pET(GFP)、pET(Et1)、 pET(EK)、pET(TEV) 擴增出靶蛋白 GFP、Et1 (TEL-SAM,轉錄抑制因子TEL的N端SAM結構域)、EK、TEV的基因片段,插入到載體的StuⅠ位點,通過藍白斑篩選和菌落PCR鑒定,依次得到重組質粒 pET(Sm’-GFP)、pET(MsbSm’-GFP)、 pET(Sm’-Et1)、pET(YdSm’-Et1)、pET(OdSm’-Et1)、pET(Sm’-EK)、pET(MsbSm’-EK)、pET(YdSm’-EK)、pET(OdSm’-EK)、pET(Sm’-TEV)、pET(MsbSm’-TEV)、pET(YdSm’-TEV) 和pET(OdSm’-TEV)。
通過 IPTG誘導,這些載體都獲得了很好地表達,融合表達蛋白的可溶性與其對應的非融合表達靶蛋白相比都不同程度地獲得了提高。其中,Sm’ 能輕微增強蛋白可溶性,但其各種復合超酸增溶標簽(MsbSm’、YdSm’、OdSm’) 的增溶效果則極其顯著(圖4),這與我們先前工作報道的結論一致[7-8]。
將每個高可溶性表達融合蛋白的細菌裂解物上清分別與表達 Ulp1酶的細菌裂解物上清進行體外反應,經SDS-PAGE分析,發現這些含Sm的融合蛋白都能與Ulp1很好地反應,Sm’及其各種復合標簽 (MsbSm’、YdSm’、OdSm’) 都能有效地從融合蛋白上切離,并得到相應的靶蛋白產物 (GFP、Et1、EK、TEV) (圖5),這說明Sm的N端附加蛋白序列并不影響 Ulp1對它的特異性酶切,而且也證實Ulp1酶的反應條件非常溫和[3,8],在細菌 PBS裂解上清中都有很好的活性。這種SUMO/Ulp1體外反應實驗可以用來分析不同標簽對靶蛋白的增溶作用機理[8]。另外,這類融合蛋白表達產物如果通過 His標簽親和層析純化,然后與純化的 Ulp1酶體外反應,將能夠有效地制備各種所需的靶蛋白 (尤其是含天然氨基酸序列的功能蛋白),與我們以前報道的脂聯素結果一致[15]。
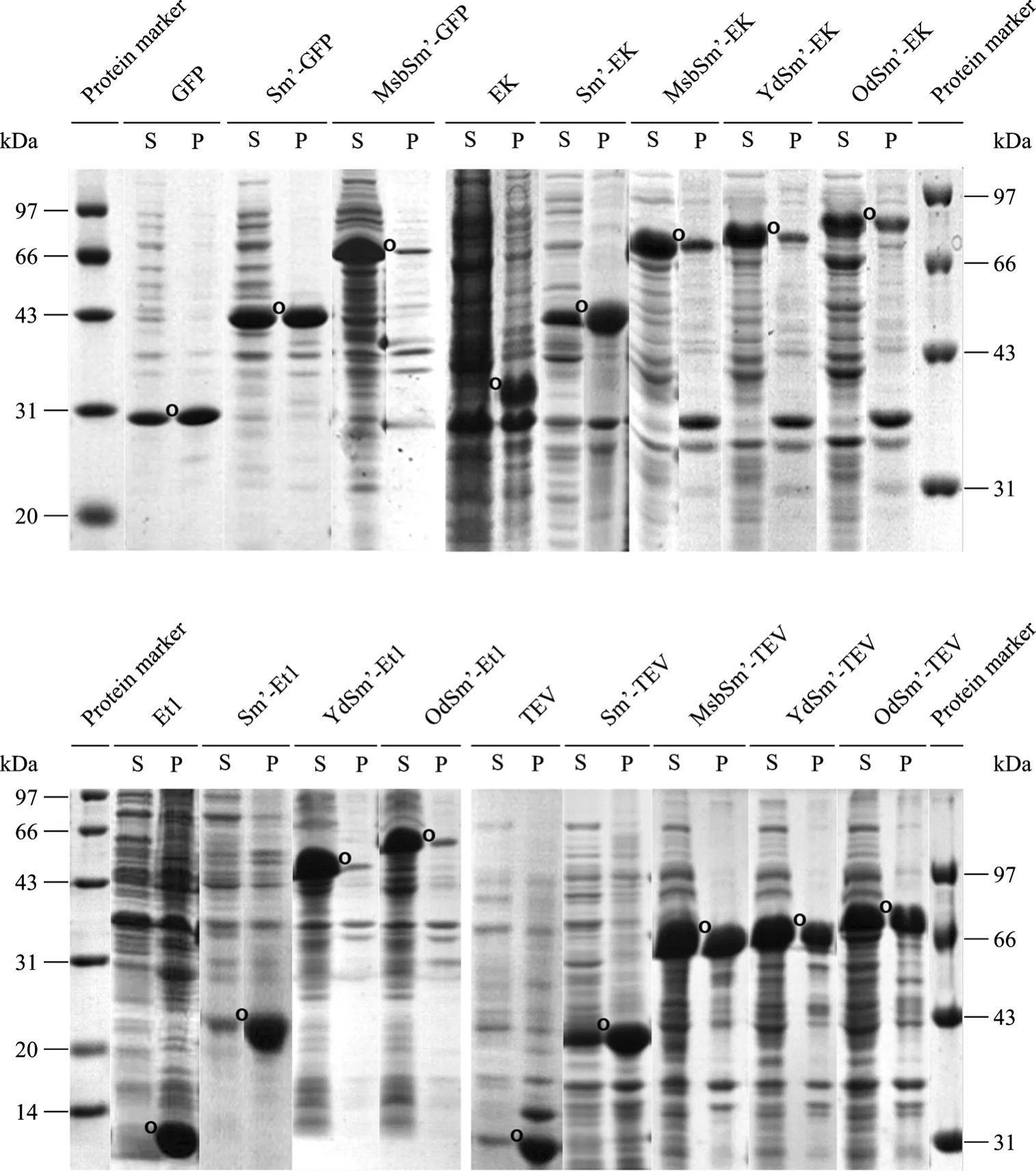
圖4 SDS-PAGE分析各種表達載體在大腸桿菌BL21(DE3) 中的表達Fig. 4 Expression of various constructs in E. coli BL21(DE3) analyzed by SDS-PAGE. O indicates the expressed target protein in non-fusion or fusion form; S: supernatant of the cell lysate; P: pellet of the cell lysate.
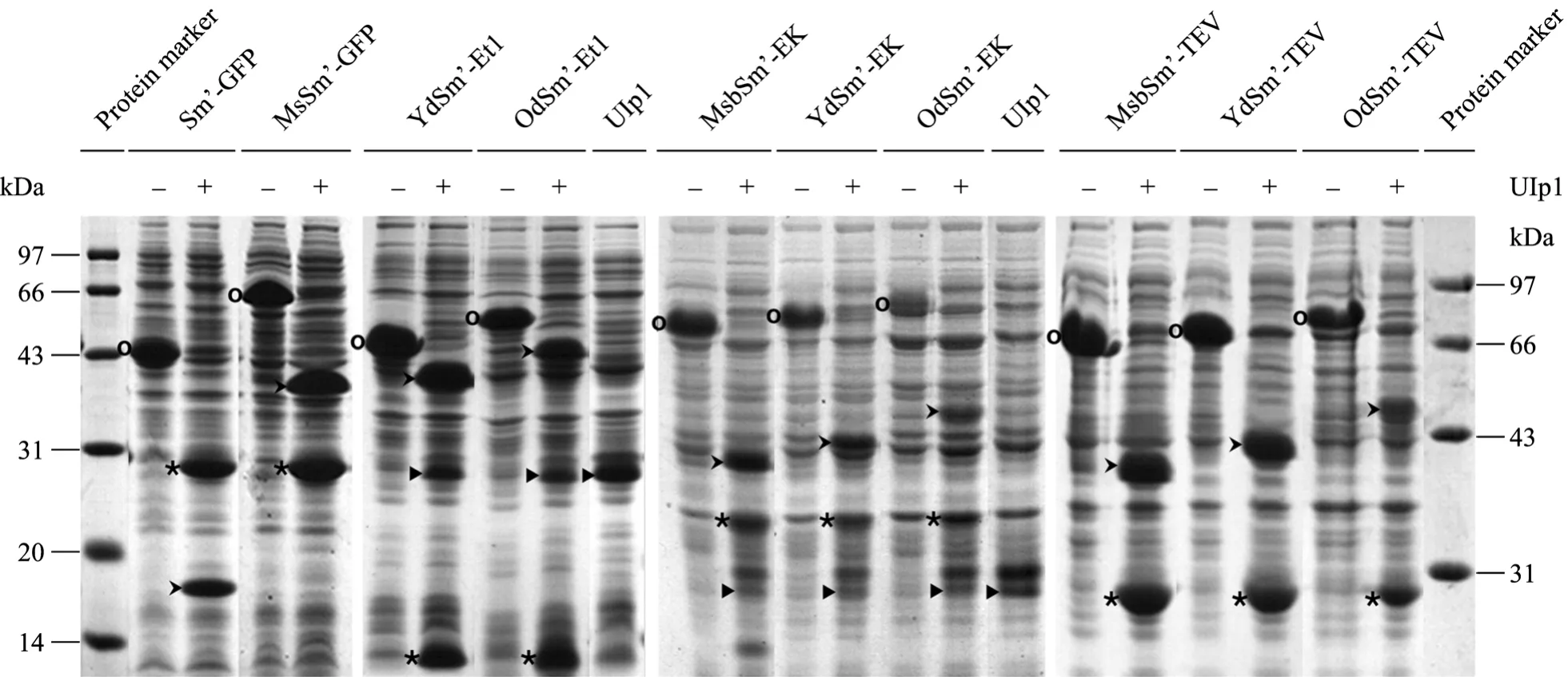
圖5 SDS-PAGE分析Sm融合蛋白與Ulp1表達細菌的裂解物上清體外酶切反應Fig. 5 In vitro enzymatic reactions of the supernatants of cell lysates of Sm fusion proteins and Ulp1, analyzed by SDS-PAGE. Oindicates Sm fusion protein; * indicates target protein; > indicates Sm’ or its combinatorial tag (e.g. YdSm’); indicates Ulp1 overused in some reactions.
2.5 基于 pAY(Sm’-LacZα) 的基因表達載體構建及其蛋白表達分析
基于上述 pAY(Sm’-LacZα) 的特性,根據需要可以快速簡便地將不同目的基因片段克隆到該質粒的MCS上 (圖3B),通過藍白斑篩選和菌落PCR鑒定得到重組質粒。
當需要誘導型表達時,可以將目的基因表達盒 (誘導型啟動子+目的基因+轉錄終止子) 插入到MCS上的任何單一酶切位點。本實驗利用引物對Pt7Fw/Tt7Rv,分別以pET(Et1)、pET(Et1-GFP)、pET(YA)、pET(MST) 為模板,擴增其對應基因表達盒片段Pt7_Et1_Tt7、Pt7_Et1-GFP_Tt7、Pt7_YA_Tt7、Pt7_MST_Tt7 (Pt7為t7啟動子,Tt7為t7轉錄終止子,位于pET載體上,圖3A)。將這些PCR片段分別平末端克隆在pAY(Sm’-LacZα) 的MCS內HincⅡ位點 (可以選擇其他平末端酶位點如 StuⅠ、SmaⅠ和Ecl136Ⅱ),構建成載體pAY(Et1)、pAY(Et1-GFP)、pAY(YA) 和pAY(MST)。如同pET系列載體一樣,它們都是受IPTG誘導表達。在不加IPTG的情況下,Pt7不會發生轉錄,同時由插入片段上游Sm啟動子Psm起始的轉錄遇到 Pt7后 LacO序列時也會被阻斷,因而沒有目的基因表達產物;當IPTG誘導時,這些pAY載體都能很好地表達 (圖6)。由于pAY載體與 pET載體使用不同的復制起始區 (p15A、ColEI),因而能夠相容于同一細菌中共表達。分別將pET(EY)/pAY(Et1)、 pET(EY)/pAY(Et1-GFP)、pET(Trx-Ab)/pAY(YA)、pET(Ab-GFP)/pAY(YA)、pET(TEV)/pAY(MST) 進行共表達,發現IPTG誘導后所有目的基因都不同程度地獲得高水平表達 (圖6),這種共表達實驗可以用來研究兩 (幾) 個關聯蛋白在細胞內的相互作用。此外,所有pAY載體在任何情況下都有一條明顯的 Cm抗性基因 (CmR) 表達條帶 (圖6),這可能與CmR上游組成型啟動子活性密切相關。
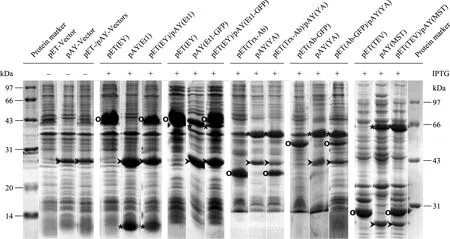
圖6 SDS-PAGE分析派生于pAY(Sm’-LacZα) 的pAY系列載體與其相關pET載體的共表達Fig. 6 Co-expressions of pAY (Sm’-LacZα)-derived pAY vectors and their correlated pET vectors, analyzed by SDS-PAGE. O indicates the recombinant protein expressed from pET vector; * indicates the recombinant protein expressed from pAY vector; > indicates the protein product of CmR (chloramphenicol acetyltransferase).
當需要組成型表達時,可以將目的基因編碼區插入在pAY(Sm’-LacZα) MCS的NcoⅠ后面任何單一酶切位點 (圖3B),并使基因翻譯閱讀框從NcoⅠ上的 ATG密碼子開始,這樣目的基因就可以由Sm啟動子Psm組成型轉錄,并由RBS(sm) 和其后NcoⅠ上 ATG密碼子介導翻譯 (圖1)。在一些細菌體內共表達分析系統中,某種蛋白 (如工具酶) 并不需要高水平的表達,這樣有必要選擇普通啟動子而不是強啟動子如 Pt7。基于此,我們利用引物對Ulp1Fw/ Ulp1Rv從 pET(Ulp1) 模板中擴增出 Ulp1片段,插入到pAY(Sm’-LacZα) 的HincⅡ位點,構建了 Ulp1組成型表達載體 pAY(c-Ulp1) (c-代表由Sm啟動子介導的組成型表達),并與上述含Sm標簽的pET系列載體如pET(YdSm’-Et1) 共表達,體內分析標簽對靶蛋白的作用,得到的結果 (未顯示) 與我們先前報道的一致[8]。
3 討論
近年來,SUMO融合技術在大腸桿菌表達系統中得到越來越多的應用,但仍處在不斷的改進中[3-8]。在本實驗中,我們發現酵母SUMO基因Smt3 (Sm)具有組成型原核啟動子活性,包含明顯的?35、?10特征序列 (圖1)。而且,通過軟莓BPROM程序預測發現,大多數物種SUMO基因編碼區普遍都具有依賴σ70的原核啟動子,但活性存在較大差異 (圖2),這種現象可能與SUMO基因作為直向同源基因(Ortholog) 在不同類物種的進化程度相關。
利用SUMO標簽本身特點及Sm內含原核啟動子特性,通過在Sm 3′末端 (-GG基序密碼子處) 引入StuⅠ位點和進一步在Sm的5′端附加His標簽和超酸增溶標簽 (Msb、Yd、Od),我們構建了基于Sm’-LacZα融合基因和 pET32a(+) 背景的系列通用克隆表達載體 (圖3A),并由多個基因克隆和表達實驗得到檢驗 (圖 4、5)。由此組成的改良 SUMO融合系統集成了諸多特性,如通過藍白斑篩選快速構建基因表達載體、SUMO和超酸增溶標簽促進蛋白可溶性表達、SUMO/Ulp1反應系統制備任何含天然氨基酸序列的靶蛋白、利用His標簽純化表達蛋白、以及pET系統固有的蛋白高水平表達等等,因此可以成為大腸桿菌重組蛋白表達的有力工具;與另一改良的SUMO融合系統[6]相比,在某些方面可能更具優勢。
另外,我們還構建了基于Sm’-LacZα融合基因、派生于質粒pACYC184 (低拷貝、含p15A復制起始區) 的通用克隆表達載體pAY(Sm’-LacZα) (圖3B)。通過藍白斑篩選鑒定,由此按需構建的pAY系列目的基因表達載體可以進行單獨表達或與其他含不同復制起始區 (如ColEI) 的質粒表達載體如pET系列共表達 (圖 6)、組成型或誘導型表達,以用于蛋白研究的不同用途如分析細胞內蛋白間的相互作用關系[8]等。
[1] Hay RT. SUMO: a history of modification. Mol Cell, 2005, 18(1): 1?12.
[2] Johnson ES. Protein modification by SUMO. Annu Rev Biochem, 2004, 73: 355?382.
[3] Malakhov MP, Mattern MR, Malakhova OA, et al. SUMO fusions and SUMO-specific protease for efficient expression and purification of proteins. J Struct Funct Genomics, 2004, 5(1/2): 75?86.
[4] Butt TR, Edavettal SC, Hall JP, et al. SUMO fusion technology for difficult-to-express proteins. Protein Expr Purif, 2005, 43(1): 1?9.
[5] Marblestone JG, Edavettal SC, Lim Y, et al. Comparison of SUMO fusion technology with traditional gene fusion systems: Enhanced expression and solubility with SUMO. Protein Sci, 2006, 15(1): 182?189.
[6] Lee CD, Sun HC, Hu SM, et al. An improved SUMO fusion protein system for effective production of native proteins. Protein Sci, 2008, 17(7): 1241?1248.
[7] Su Y, Zou ZR, Feng SY, et al. The acidity of protein fusion partners predominantly determines the efficacy to improve the solubility of the target proteins expressed in Escherichia coli. J Biotech, 2007, 129(3): 373?382.
[8] Zou ZR, Cao LJ, Zhou P, et al. Hyper-acidic protein fusion partners improve solubility and assist correct folding of recombinant proteins expressed in Escherichia coli. J Biotech, 2008, 135(4): 333?339.
[9] Mossessova E, Lima CD. Ulp1-SUMO crystal structure and genetic analysis reveal conserved interactions and a regulatory element essential for cell growth in yeast. Mol Cell, 2000, 5(5): 865?876.
[10] Catanzariti AM, Soboleva TA, Jans DA, et al. An efficient system for high-level expression and easy purification of authentic recombinant proteins. Protein Sci, 2004, 13(5): 1331?1339.
[11] Catic A, Misaghi S, Korbel GA, et al. ElaD, a deubiquitinating protease expressed by E. coli. PLoS ONE, 2007, 2(4): e381.
[12] Sambrook J, Russell DW. Molecular Cloning: A Laboratory Manual. 3rd Ed. New York: Cold Spring Harbor Laboratory Press, 2001.
[13] Horton RM, Hunt HD, Ho SN, et al. Engineering hybrid genes without the use of restriction enzymes: gene splicing by overlap extension. Gene, 1989, 77(1): 61?68.
[14] Rose RE. The nucleotide sequence of pACYC184. Nucleic Acids Res, 1988, 16(1): 355.
[15] Zhou P, Feng SY, Zou ZR, et al. Soluble expression of human adiponectin in Escherichia coli. J Sichuan Univ: Nat Sci Ed, 2010, 47(1): 161?166.周培, 封淑穎, 鄒竹榮, 等. 人脂聯素在大腸桿菌中的可溶性表達. 四川大學學報:自然科學版, 2010, 47(1): 161?166.
猜你喜歡
中學生數理化·中考版(2022年8期)2022-06-14 06:55:24
數學年刊A輯(中文版)(2022年4期)2022-02-16 08:17:34
今日農業(2021年19期)2022-01-12 06:16:36
中老年保健(2021年11期)2021-08-22 03:15:44
無線電通信技術(2021年4期)2021-07-13 08:58:28
無線電通信技術(2021年3期)2021-06-08 03:33:48
中學生數理化(高中版.高考數學)(2021年1期)2021-03-19 08:28:38
無線電工程(2020年11期)2020-10-29 01:25:46
現代出版(2020年3期)2020-06-20 07:10:34
福利中國(2015年4期)2015-01-03 08:03:38
