中央核肌病1 例報道
2010-11-20 05:21:16段維維李秋香梁靜慧
卒中與神經疾病 2010年6期
段維維 李秋香 梁靜慧 楊 歡
1 病 例
患者女性,13 歲, 因雙下肢無力11 年就診。患者1 歲半時學會走路,家長發現其步態不穩, 易跌倒,跌倒后尚能自行站起,逐漸出現上下樓梯費力, 蹲下起立需扶助, 后出現穿拖鞋易掉落, 尚能行走, 但較慢,不能進行體育活動;肢體無麻木疼痛、肉跳感, 無吞咽困難、飲水嗆咳等。出生史及既往史無特殊。家族中無類似患者, 父母非近親婚配。飲食、睡眠和大小便均正常。
體格檢查:營養中等,發育較同齡人無明顯差異,輕度鴨步,雙上肢肌力正常,雙下肢近端肌力4 級, 遠端2 級, 足背屈不能,Gowers 征(+),深淺感覺正常, 四肢肌張力偏低, 下肢肌肉輕度萎縮。四肢腱反射對稱性減低, 病理征未引出。血清肌酶正常。肌電圖示肌源性損害。
分析:此患者表現為下運動單位受損:肌無力、肌萎縮,無感覺障礙,病理征(-), 肌電圖呈肌源性損害, 血清肌酶學正常。定位診斷:肌肉疾病。定性診斷:先天性肌病? 先天性肌營養不良? 進一步行骨骼肌活檢病理檢查, 取腓腸肌適量肌纖維標本送檢, 常規組織化學、酶學染色, H E 染色可見細胞核位于肌細胞中央,多核或雙核(圖1);ATP 染色顯示I型肌纖維為主(圖2)。結合臨床, 診斷先天性肌病-中央核肌病(centronuclear myopathy,CNM)。
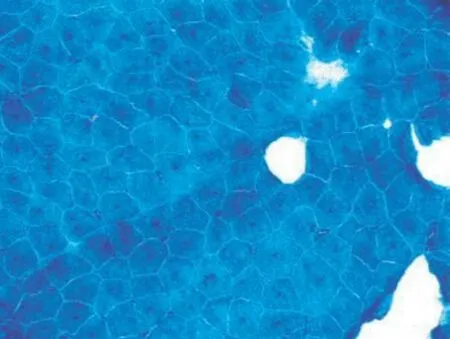
圖2 ATP 染色以I 型肌纖維為主
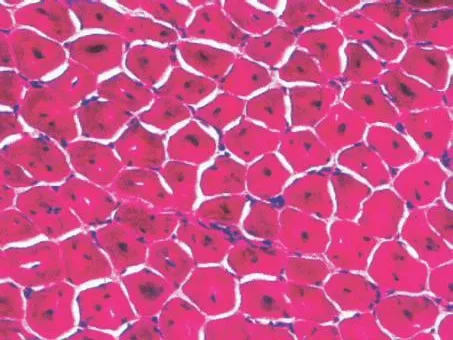
圖1 H E 染色可見細胞核位于肌細胞中央、多核或雙核
2 討 論
中央核肌病是一種少見的先天性肌肉疾病, 由Spiro 等于1966 年首先報道, 因其病理形態學特點為單個的肌核出現在許多肌纖維的中央, 和發育期的肌管非常相似, 故又被稱為肌管肌病(M TM)。目前尚無有效治療方法。中央核肌病是一組從臨床表現到遺傳方式都呈明顯異質性的肌病。Heckmatt 等依據其臨床表現和遺傳方式將其分為(1)X 連鎖隱性遺傳新生兒或新生兒前期發病的嚴重型;(2)常染色體隱性遺傳嬰兒期或兒童期發病型;(3)兒童晚期至成人期發病的常染色體顯性遺傳型。 尚有散發病例報道。 在這些類型中X 連鎖隱性遺傳型是由于肌微管素(myotubularin,MTM1)基因突變所致, 其位點定位于Xq28。該型新生兒期即開始出現明顯的肌無力肌張力低下, 眼外肌麻痹及呼吸困難,多于1 歲前死亡, 常伴有巨顱、狹長臉型及手指、腳趾細長,這可能有助于診斷。常染色體顯性遺傳型, 其進展緩慢,臨床表現相對較輕, 與動力蛋白2 基因(dynamin 2 gene,DNM2)的突變有關。而常染色體隱性遺傳型, 基因定位不明,其臨床嚴重程度介于前兩者之間。常表現為面肌受累,如咀嚼肌無力、眼瞼下垂、眼外肌麻痹等。其肌無力以近端多見,也可有遠端肌的無力與萎縮,足畸形也較常見。根據該患者的臨床資料,其分型尚不明確。
本病的確診尚有賴于肌肉病檢。中央核肌病的病理表現為細胞核排列于肌纖維中心(中心核纖維), 幾乎所有病例中都有I 型肌纖維優勢和萎縮。 骨骼肌形成時單核的成肌細胞聚集,互相融合而成多核的細長細胞, 這種融合細胞中心部有核,稱為肌管細胞。成肌細胞內肌原纖維規則排列,接受神經支配后才稱為肌纖維。 肌管性肌病的骨骼肌纖維細長,與肌管細胞非常相似, 所以稱為肌管性肌病。 肌纖維接受神經的支配,分化良好, 但并不一定反映肌管細胞停止成熟,因此稱為中央核肌病更恰當。
臨床表現肌力、肌張力低下,血CK 正常或輕度升高, 肌電圖呈肌源性損害, 臨床考慮肌病, 必須進一步行開放式骨骼肌活檢才能明確診斷和鑒別診斷。此病病理特點為細胞核位于肌細胞中央,呈多核或雙核。因中心核自身并無特殊性,可見于多種病理過程, 如失神經支配、肌營養不良、代謝性肌病、中毒性肌病、再生纖維等,因此本病的診斷需結合遺傳學特征、臨床表現綜合判斷。
