色譜保留時(shí)間在蛋白質(zhì)組研究中的應(yīng)用
2010-10-21 03:47:24高友鶴
色譜 2010年2期
關(guān)鍵詞:實(shí)驗(yàn)方法
邵 晨, 高友鶴
(中國(guó)醫(yī)學(xué)科學(xué)院基礎(chǔ)醫(yī)學(xué)研究所,中國(guó)協(xié)和醫(yī)科大學(xué)基礎(chǔ)醫(yī)學(xué)院,生理和病理生理學(xué)系,北京100005)
色譜保留時(shí)間在蛋白質(zhì)組研究中的應(yīng)用
邵 晨*, 高友鶴
(中國(guó)醫(yī)學(xué)科學(xué)院基礎(chǔ)醫(yī)學(xué)研究所,中國(guó)協(xié)和醫(yī)科大學(xué)基礎(chǔ)醫(yī)學(xué)院,生理和病理生理學(xué)系,北京100005)
液相色譜與串聯(lián)質(zhì)譜聯(lián)用(LC-MS/MS)技術(shù)是蛋白質(zhì)組學(xué)研究中的常見方法。保留時(shí)間作為獨(dú)立于質(zhì)譜信息的參數(shù)已經(jīng)被用于蛋白質(zhì)的鑒定和定量工作中。在多肽鑒定領(lǐng)域,多肽的色譜保留時(shí)間預(yù)測(cè)與常規(guī)的二級(jí)串聯(lián)質(zhì)譜數(shù)據(jù)庫(kù)搜索算法結(jié)合可以提高鑒定的可信度。鑒定的靈敏度也可以通過(guò)匹配多次LC-MS實(shí)驗(yàn)中具有相同精確質(zhì)量數(shù)和保留時(shí)間的峰而提高。另一方面,由于色譜條件的微小改變即會(huì)引起保留時(shí)間的變化,因此對(duì)多次實(shí)驗(yàn)結(jié)果進(jìn)行保留時(shí)間比對(duì)是進(jìn)行非標(biāo)記定量的不可或缺的步驟。另外,聯(lián)合保留時(shí)間偏移和質(zhì)量數(shù)信息還可以進(jìn)行蛋白質(zhì)翻譯后修飾(post-translational modification,PTM)的鑒定。
液相色譜-串聯(lián)質(zhì)譜;保留時(shí)間預(yù)測(cè);保留時(shí)間比對(duì);多肽鑒定;非標(biāo)記定量;翻譯后修飾;蛋白質(zhì)組學(xué)
Abstract:Liquid Chromatography coup led with tandem mass spectrometry(LC-MS/MS)has been one of the most popular approaches inproteome analysis.As an independent parameter to mass spectrometry information,peptide retention time has been utilized to facilitate protein identification and quantification.In the field of pep tide identification,the prediction of the retention time combined with routine tandem mass spectrometry database searching methods could help improve the confidence of identification.The sensitivity of identification could also be improved by matching peaks with both the accurate mass and retention time in multiple aligned LC-MS runs.Meanwhile,because small changes of liquid Chromatography conditions lead to variability in retention times unavoidably,retention time alignm ent is crucial to label free quantification.Additionally,post-translational modifications(PTM)could be identified by com bining retention time shifts and mass deviation information.
Key words:liquid Chromatography-tandem mass spectrometry(LC-MS/MS);retention time prediction;retention time alignment;peptideidentification;label-free quantification;posttranslational modification(PTM);proteomics
在蛋白質(zhì)組的研究中,液相色譜串聯(lián)一級(jí)質(zhì)譜(liquid Chromatography coupled with mass spectrom etry,LC-MS)或二級(jí)質(zhì)譜(liquid Chromatography coup led with tandem mass spectrometry,LC-MS/MS)是進(jìn)行蛋白質(zhì)鑒定和定量分析的常見策略。其主要實(shí)驗(yàn)流程為:首先將蛋白質(zhì)混合物經(jīng)酶切變?yōu)槎嚯幕旌衔?然后通過(guò)一維或二維色譜分離多肽混合物,再用質(zhì)譜儀鑒定其序列及進(jìn)行定量分析。用一維色譜分離多肽混合物時(shí),通常是利用反相液相色譜(reversed-phase liquid Chromatography,RPLC)根據(jù)多肽的疏水性進(jìn)行分離,或者根據(jù)多肽的電荷特性在RPLC之前再加上強(qiáng)陽(yáng)離子交換(strong cation exchange,SCX)色譜進(jìn)行二維分離。在蛋白質(zhì)組的鑒定和定量分析中,有相當(dāng)一部分利用了保留時(shí)間的信息。本文將對(duì)保留時(shí)間的預(yù)測(cè)算法和比對(duì)算法及其在蛋白質(zhì)組研究中的應(yīng)用進(jìn)行綜述。
我們將首先介紹兩項(xiàng)主要技術(shù)——保留時(shí)間的預(yù)測(cè)和保留時(shí)間的比對(duì)——的最新研究進(jìn)展。隨后,將介紹以這兩項(xiàng)技術(shù)為基礎(chǔ)的一系列蛋白質(zhì)組數(shù)據(jù)分析算法,包括多肽序列鑒定、翻譯后修飾(post-translational m odification,PTM)鑒定和非標(biāo)記定量。
1 保留時(shí)間的預(yù)測(cè)
以往大多數(shù)的蛋白質(zhì)組鑒定工作都是由質(zhì)譜的數(shù)據(jù)出發(fā),比如利用肽段母離子質(zhì)荷比(mass-tocharge ratio,m/z)和MS/MS譜圖中的碎片離子信息等。但是由于多肽混合物的復(fù)雜性和噪聲信號(hào)的影響,鑒定中存在著假陽(yáng)性和假陰性。多肽的保留時(shí)間在近幾年被應(yīng)用到質(zhì)譜的鑒定中。理想狀況下,當(dāng)色譜分離條件(溫度、pH值、流動(dòng)相組成和固定相)固定時(shí),多肽的保留時(shí)間也應(yīng)保持不變。由于大部分的多肽都不具有實(shí)驗(yàn)獲得的保留時(shí)間信息,所以預(yù)測(cè)保留時(shí)間成為一個(gè)很好的替代方法。進(jìn)行保留時(shí)間預(yù)測(cè)的理論根據(jù)是多肽在色譜中的行為與其序列、結(jié)構(gòu)和物理化學(xué)性質(zhì)相關(guān)。這里將介紹主要的保留時(shí)間預(yù)測(cè)方法。由于這些預(yù)測(cè)方法的效果大多是用預(yù)測(cè)與實(shí)際測(cè)定的保留時(shí)間的相關(guān)關(guān)系來(lái)表示,而相關(guān)系數(shù)的大小強(qiáng)烈地依賴于進(jìn)行驗(yàn)證的數(shù)據(jù),因此很難通過(guò)文獻(xiàn)報(bào)道比較這些方法的預(yù)測(cè)效果的好壞。
1.1 根據(jù)多肽的序列預(yù)測(cè)保留時(shí)間
最簡(jiǎn)單的保留時(shí)間預(yù)測(cè)方法是估計(jì)每一個(gè)氨基酸殘基對(duì)保留時(shí)間的貢獻(xiàn)值(系數(shù)),再根據(jù)多肽的氨基酸組成將它們的保留時(shí)間系數(shù)加和在一起。這種方法是建立在多肽的保留時(shí)間主要是由其氨基酸組成所決定這一假設(shè)的基礎(chǔ)上的。一種估計(jì)氨基酸保留時(shí)間貢獻(xiàn)的方法[1]是合成特定序列的多肽Ac-G ly-X-X-(Leu)3-(Lys)2-am ide(X為任意20個(gè)氨基酸之一),通過(guò)測(cè)定這些多肽在RPLC的保留時(shí)間,從而計(jì)算出每一個(gè)氨基酸的保留時(shí)間(疏水性)系數(shù)。在隨后的研究中,同一個(gè)研究組又引入了一個(gè)校正因子,以消除肽段長(zhǎng)度對(duì)保留時(shí)間的影響[2]。除了合成多肽外,很多研究組都利用線性回歸模型[3-6],從測(cè)定到的已知序列多肽的保留時(shí)間中計(jì)算每個(gè)氨基酸殘基的保留時(shí)間系數(shù)。
近幾年,這種通過(guò)加和每個(gè)氨基酸的保留時(shí)間系數(shù)計(jì)算多肽保留時(shí)間的方法又有了新的改進(jìn)。Petritis等[7]用更加智能化的人工神經(jīng)網(wǎng)絡(luò)算法重新計(jì)算了氨基酸的保留時(shí)間參數(shù)。他們計(jì)算的新參數(shù)與Guo研究組[1,8]的結(jié)果有一定的相似性,不同的是,他們認(rèn)為亮氨酸對(duì)保留時(shí)間的影響最大,這又與B row ne等[3]的結(jié)論相同。Krokhin等[9]發(fā)現(xiàn),除了氨基酸組成和肽鏈的長(zhǎng)度外,多肽N末端的氨基酸殘基對(duì)保留時(shí)間也有很大的影響,因此在他們的預(yù)測(cè)公式中加入了關(guān)于肽段長(zhǎng)度和多肽N末端3個(gè)殘基的校正因子。Kaw akam i等[10]則研究了翻譯后修飾對(duì)保留時(shí)間的影響。他們發(fā)現(xiàn)同樣是磷酸化修飾,但磷酸化的絲氨酸延遲的保留時(shí)間最短,磷酸化的蘇氨酸的延遲時(shí)間有少許增加,而磷酸化的酪氨酸則會(huì)產(chǎn)生較長(zhǎng)時(shí)間的延遲。這一發(fā)現(xiàn)提示保留時(shí)間可以應(yīng)用于鑒定多肽的翻譯后修飾及其位點(diǎn)。
1.2 根據(jù)多肽的物理化學(xué)性質(zhì)預(yù)測(cè)保留時(shí)間
Petritis等[11]在2006年對(duì)他們以前的工作進(jìn)行了改進(jìn),仍然應(yīng)用人工神經(jīng)網(wǎng)絡(luò)算法,但考慮了多肽的物理化學(xué)性質(zhì)信息,其中包括了多肽的長(zhǎng)度、序列、氨基酸疏水性、疏水性矩、預(yù)測(cè)的二級(jí)結(jié)構(gòu)以及相鄰的氨基酸組合出現(xiàn)的頻率。新方法預(yù)測(cè)的準(zhǔn)確率得到了顯著的提高,預(yù)測(cè)與實(shí)驗(yàn)的保留時(shí)間平均誤差為1.5%。由于考慮了多肽序列信息,算法還可以成功地對(duì)蛋白質(zhì)異構(gòu)體進(jìn)行區(qū)分。
另一種通過(guò)物理化學(xué)性質(zhì)預(yù)測(cè)多肽保留時(shí)間的模型被稱作定量結(jié)構(gòu)-保留相關(guān)關(guān)系(quantitative structure retention relationship,QSRR)[12,13]。這個(gè)模型表示為:
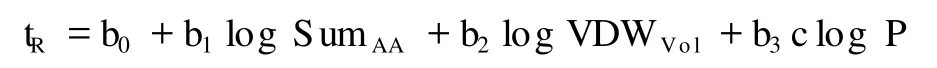
其中:tR為多肽在梯度洗脫下的保留時(shí)間;b0為一常數(shù);SumAA是全部氨基酸殘基的保留時(shí)間系數(shù)之和;VDWVol指多肽的范氏體積;clog P則是對(duì)數(shù)化的正辛醇-水分配系數(shù);b1,b2,b3為上述3個(gè)參數(shù)的權(quán)重,可利用線性回歸模型得到。
Asenjo研究組[14-17]一直致力于通過(guò)研究多肽表面的氨基酸和色譜柱的疏水性相互作用來(lái)預(yù)測(cè)保留時(shí)間。這種方法需要已知多肽結(jié)構(gòu)和建立復(fù)雜的數(shù)學(xué)模型做出相應(yīng)的預(yù)測(cè),而且預(yù)測(cè)花費(fèi)的時(shí)間較長(zhǎng),并且通量較低。2005年他們研究了一種方法可以只基于氨基酸序列進(jìn)行預(yù)測(cè)[14,15]。該方法首先通過(guò)多肽的每個(gè)氨基酸殘基最大可達(dá)到的表面積及其暴露在表面的可能性來(lái)估計(jì)多肽的表面積,再根據(jù)其相對(duì)分子質(zhì)量、物理化學(xué)性質(zhì)、二級(jí)結(jié)構(gòu)等特征,利用機(jī)器學(xué)習(xí)算法預(yù)測(cè)多肽的保留時(shí)間。
2 保留時(shí)間的比對(duì)
在蛋白質(zhì)組特別是臨床蛋白質(zhì)組的研究中,往往需要通過(guò)比較很多例的樣品來(lái)發(fā)現(xiàn)潛在的疾病標(biāo)志物或有特定含義的差異蛋白質(zhì),發(fā)現(xiàn)方法主要有標(biāo)記定量和非標(biāo)記定量。標(biāo)記定量的方法由于標(biāo)記試劑種類的限制,只能對(duì)數(shù)量有限的樣品進(jìn)行定量。而非標(biāo)記定量方法對(duì)每例樣品先分別進(jìn)行LC-MS分析,再將得到的結(jié)果先進(jìn)行保留時(shí)間比對(duì)再定量分析。由于不需要事先進(jìn)行樣品混合,非標(biāo)記定量可以進(jìn)行成百上千例樣品的定量,這就克服了標(biāo)記定量只能比較有限樣品數(shù)的缺點(diǎn)[18]。把不同次實(shí)驗(yàn)產(chǎn)生的LC-MS譜圖進(jìn)行保留時(shí)間比對(duì)是進(jìn)行非標(biāo)記定量的重要步驟。LC-MS譜圖比對(duì)不但可以消除實(shí)驗(yàn)間的色譜分離分析誤差,而且使不同時(shí)間、不同實(shí)驗(yàn)室產(chǎn)生的LC-MS結(jié)果進(jìn)行同時(shí)比較成為可能。
進(jìn)行LC-MS譜圖比對(duì)的另一個(gè)應(yīng)用是在蛋白質(zhì)鑒定方面。由于LC-MS/MS實(shí)驗(yàn)中,一級(jí)質(zhì)譜掃描到的多肽質(zhì)譜峰只有少部分會(huì)被選擇進(jìn)行二級(jí)質(zhì)譜鑒定得到序列信息,因而在單次的蛋白質(zhì)組實(shí)驗(yàn)中,大量的多肽(或者蛋白質(zhì))都不能得到鑒定。如果假設(shè)具有相同質(zhì)量數(shù)和保留時(shí)間特征的質(zhì)譜峰所代表的是同一個(gè)多肽,通過(guò)把同樣或相似樣品的多次LC-MS譜圖比對(duì)在一起,只要這個(gè)多肽在其中的一次LC-MS/MS中鑒定出來(lái),就可以把其他的實(shí)驗(yàn)中具有同樣特征的質(zhì)譜峰也鑒定為這個(gè)多肽,這就大大提高了鑒定的多肽和蛋白質(zhì)的覆蓋率和靈敏度[19]。
2.1 保留時(shí)間偏差的來(lái)源
在色譜分離過(guò)程中,相當(dāng)一部分誤差是由色譜柱本身產(chǎn)生的,主要包括色譜柱老化、填充不均勻以及柱內(nèi)殘留的污染物的影響等[20]。即使實(shí)驗(yàn)條件控制得很好,這些誤差也很難避免。另外,即使是同樣的實(shí)驗(yàn)條件和樣品,更換色譜柱也會(huì)造成色譜圖的差別。另一方面,一些色譜實(shí)驗(yàn)條件(如溫度、洗脫梯度等)很難控制也是產(chǎn)生保留時(shí)間偏差的主要原因。最后,儀器產(chǎn)生的誤差(如死體積和流速的變化、基線漂移等)也會(huì)造成很大的影響。因此,在分析不同次的LC-MS數(shù)據(jù)時(shí),進(jìn)行保留時(shí)間比對(duì)是不可或缺的步驟。
2.2 保留時(shí)間比對(duì)的算法
保留時(shí)間比對(duì)的算法大致可分為兩種:一種是全譜比對(duì)算法,即對(duì)整個(gè)未處理的LC圖譜進(jìn)行全局比對(duì),而幾乎不考慮質(zhì)譜的信息;另一種方法則只比對(duì)從總LC-MS譜圖中提取出的可能代表多肽的質(zhì)譜峰,需要將峰的m/z列入計(jì)算。由于在比對(duì)時(shí)只保留了有意義的質(zhì)譜峰,第二種方法需要計(jì)算的數(shù)據(jù)量較小,但比對(duì)的結(jié)果非常依賴于多肽峰的檢測(cè)算法。總的來(lái)講,前一種方法能夠處理低m/z分辨率的數(shù)據(jù),但計(jì)算量較大,不適宜進(jìn)行過(guò)多的LCMS數(shù)據(jù)的同時(shí)比較;而后一種方法往往需要精確的質(zhì)量數(shù)來(lái)判斷不同實(shí)驗(yàn)間代表同一多肽的質(zhì)譜峰,對(duì)質(zhì)譜儀的要求較高。
2.2.1 LC全譜比對(duì)算法
全譜比對(duì)算法主要比對(duì)不同次LC-MS的總離子流(total ion current,TIC)色譜圖,一次實(shí)驗(yàn)的TIC譜圖可以視為一條在不同的時(shí)間點(diǎn)具有不同的總離子流量的曲線。這種曲線在數(shù)學(xué)上稱為連續(xù)時(shí)間序列。這樣,進(jìn)行兩次實(shí)驗(yàn)間比對(duì)的任務(wù)就可以歸納為這樣一個(gè)數(shù)學(xué)問(wèn)題:尋找一個(gè)轉(zhuǎn)換函數(shù),使兩條曲線之間的距離最小。根據(jù)對(duì)曲線間距離大小的不同定義,可以用多種動(dòng)態(tài)規(guī)劃算法,如動(dòng)態(tài)時(shí)間規(guī)整(dynam ic time w arp ing,D TW)[21]、相關(guān)優(yōu)化偏移(correlation op tim ized w arp ing,COW)[22]、參數(shù)時(shí)間規(guī)整(param etric time w arp ing,PTW)[23]等來(lái)求解轉(zhuǎn)換函數(shù)。
除了完全基于TIC譜圖的方法以外,也有一些算法利用了質(zhì)譜的信息,即先將總的TIC譜圖分成在不同的m/z區(qū)間內(nèi)的子譜圖,再進(jìn)行比對(duì)打分。這樣可以對(duì)復(fù)雜度更高的樣品進(jìn)行較好的比對(duì)。Listgarten等[24]在使用隱馬氏模型進(jìn)行比對(duì)時(shí),發(fā)現(xiàn)把每個(gè)保留時(shí)間點(diǎn)的總離子流量分入4個(gè)m/z區(qū)間時(shí),既可以提高比對(duì)的精確度,也不會(huì)帶來(lái)過(guò)大的計(jì)算負(fù)擔(dān)。
2.2.2 多肽特征峰比對(duì)算法
對(duì)TIC譜圖進(jìn)行比對(duì)只適合樣品的復(fù)雜度比較低的情況。當(dāng)混合物的復(fù)雜度較高時(shí),不同的多肽可能在同一時(shí)間流出,其色譜峰重疊在一起,在TIC譜圖中不能區(qū)分。在蛋白質(zhì)組的研究中,實(shí)際關(guān)心的只是代表多肽的質(zhì)譜信號(hào)。多肽特征峰比對(duì)算法首先檢測(cè)LC-MS譜圖中可能是多肽的質(zhì)譜峰(可通過(guò)具有較高的信噪比,或經(jīng)由MS/MS鑒定得到序列等特征來(lái)判斷),稱為特征峰(feature),再將可能代表同一多肽的質(zhì)譜峰匹配起來(lái),比對(duì)的目標(biāo)是使同一多肽的保留時(shí)間在歷次實(shí)驗(yàn)間的誤差最小。利用MS/MS的鑒定結(jié)果進(jìn)行特征峰判定最為可靠,但是MS/MS數(shù)據(jù)不易獲得。由于MS/MS掃描速率較慢,在全部可能是多肽的質(zhì)譜峰中,僅有少部分具有MS/MS鑒定結(jié)果。M ueller等[25]報(bào)道95%的質(zhì)譜峰用MS/MS鑒定都可被判定為特征峰,但在他們提取的全部特征峰中僅有10%進(jìn)行了MS/MS鑒定。
在M ueller小組的方法中,落在相近的m/z和保留時(shí)間范圍內(nèi)的特征峰被分為一組,采用局部加權(quán)回歸散點(diǎn)平滑法估計(jì)兩次實(shí)驗(yàn)間同一組特征峰保留時(shí)間的變化。與M ueller等比對(duì)全部特征峰的方法相反,Petritis等[7]只選擇了6個(gè)在多次實(shí)驗(yàn)中經(jīng)常被MS/MS鑒定出序列的多肽作為比對(duì)的依據(jù)。他們采用遺傳算法計(jì)算每次實(shí)驗(yàn)的線性保留時(shí)間轉(zhuǎn)換函數(shù),同時(shí)對(duì)多次實(shí)驗(yàn)的結(jié)果進(jìn)行比對(duì),最后將保留時(shí)間歸一化到0~1的區(qū)間里。
Fischer等[26]采取了折中的辦法,首先用嶺回歸算法根據(jù)高可信度的MS/MS數(shù)據(jù)對(duì)兩次實(shí)驗(yàn)數(shù)據(jù)進(jìn)行第一次比對(duì),計(jì)算得到一個(gè)多項(xiàng)式作為保留時(shí)間轉(zhuǎn)換函數(shù)。在初始比對(duì)的結(jié)果上,找到相關(guān)度最高的未經(jīng)MS/MS鑒定的特征峰,然后根據(jù)它們的保留時(shí)間偏差對(duì)多項(xiàng)式進(jìn)行修正,如此經(jīng)過(guò)數(shù)輪迭代,可獲得最佳的比對(duì)效果。2007年,該小組在原來(lái)的算法上做了進(jìn)一步改進(jìn)[27],利用多元典型相關(guān)分析替代嶺回歸,解決了原來(lái)的算法不具有對(duì)稱性的問(wèn)題(對(duì)稱是指將LC-MS譜圖A比對(duì)到譜圖B上和將譜圖B比對(duì)到譜圖A上獲得的結(jié)果相同)。
2.2.3 選擇恰當(dāng)?shù)谋葘?duì)算法
最簡(jiǎn)單的保留時(shí)間比對(duì)是僅僅通過(guò)線性回歸來(lái)校正不同次實(shí)驗(yàn)間的保留時(shí)間變化,這種方法雖然比較粗糙,但計(jì)算速度最快,健壯性較好。一般情況下,當(dāng)色譜條件完全相同時(shí),實(shí)驗(yàn)間的誤差用線性變換來(lái)校正即可。然而,大多數(shù)的實(shí)驗(yàn)都存在著非線性的保留時(shí)間誤差。計(jì)算非線性的保留時(shí)間轉(zhuǎn)換函數(shù)不僅可以應(yīng)對(duì)洗脫梯度不同的情況,而且比對(duì)更為精確。Podw ojski等[28]比較了線性回歸方法和兩種非線性轉(zhuǎn)換函數(shù),肯定了在對(duì)比對(duì)的精度要求較高時(shí)使用非線性轉(zhuǎn)換函數(shù)的必要性。但是非線性算法不僅對(duì)計(jì)算機(jī)的要求較高,轉(zhuǎn)換函數(shù)過(guò)于復(fù)雜時(shí)還存在著過(guò)擬合的風(fēng)險(xiǎn),因此需要謹(jǐn)慎地選擇。Vandenbogaert等[20]建議首先選擇任意一個(gè)可以進(jìn)行非線性比對(duì)的軟件,用它的比對(duì)結(jié)果來(lái)判斷數(shù)據(jù)是否具有非線性的保留時(shí)間誤差,再選擇恰當(dāng)?shù)耐燃?jí)(線性或非線性)的算法進(jìn)行比對(duì)。
不同的比對(duì)算法對(duì)LC-MS數(shù)據(jù)本身也有不同的要求。TIC譜圖比對(duì)需要的計(jì)算量最大,當(dāng)需要比對(duì)的實(shí)驗(yàn)次數(shù)過(guò)多或混合物的復(fù)雜度較高時(shí),不適宜使用這種算法。特征峰比對(duì)算法首先要進(jìn)行特征提取的步驟,加入這一步驟也帶來(lái)了額外的誤差,尤其是低分辨率質(zhì)譜儀產(chǎn)生的數(shù)據(jù),在進(jìn)行特征峰提取時(shí)將產(chǎn)生較大的誤差,對(duì)比對(duì)的結(jié)果有很大的影響。
除了數(shù)據(jù)本身的特征以外,還應(yīng)根據(jù)比對(duì)的目的來(lái)選擇比對(duì)算法。如果是通過(guò)比較蛋白質(zhì)組發(fā)現(xiàn)疾病標(biāo)志物的工作,可以只對(duì)保留時(shí)間進(jìn)行較粗略的全局校正以方便定量;而如果是通過(guò)保留時(shí)間和精確質(zhì)量數(shù)鑒定多肽序列的工作,則對(duì)比對(duì)的精度要求很高。總而言之,在選擇保留時(shí)間比對(duì)算法時(shí),應(yīng)根據(jù)數(shù)據(jù)的特征和實(shí)際應(yīng)用的需要,選擇最適合而不是最精密復(fù)雜的算法,在比對(duì)精度、算法健壯性和計(jì)算時(shí)間之間獲得最佳的平衡。
3 利用保留時(shí)間進(jìn)行多肽鑒定
3.1 利用精確質(zhì)量數(shù)和保留時(shí)間根據(jù)一級(jí)質(zhì)譜數(shù)據(jù)鑒定多肽
隨著質(zhì)譜技術(shù)的發(fā)展和質(zhì)譜儀精度的提高,一些研究試圖僅利用酶切多肽的精確質(zhì)量數(shù)和色譜的保留時(shí)間鑒定多肽的序列,即只進(jìn)行LC-MS實(shí)驗(yàn),而不需要再做二級(jí)質(zhì)譜分析。這種方法通常是針對(duì)某一特定的組織,通過(guò)收集多次LC-MS/MS實(shí)驗(yàn)的鑒定結(jié)果,建立起這一特定組織的多肽質(zhì)量和保留時(shí)間標(biāo)簽(accurate mass and time tag,AM T tag)數(shù)據(jù)庫(kù)。在隨后對(duì)該組織的實(shí)驗(yàn)中,只進(jìn)行LCMS,而不用二級(jí)質(zhì)譜分析,就可以通過(guò)搜索之前建立的數(shù)據(jù)庫(kù)來(lái)鑒定多肽序列。這種鑒定方法可以大幅度地節(jié)約進(jìn)行二級(jí)質(zhì)譜的時(shí)間。由于并不是一級(jí)質(zhì)譜的每一個(gè)峰都被選擇進(jìn)行二級(jí)質(zhì)譜分析,而且只有質(zhì)量好的二級(jí)質(zhì)譜譜圖中的多肽會(huì)得到正確鑒定,LC-MS/MS的鑒定方法產(chǎn)生了大量的假陰性,且靈敏度不夠。只利用LC-MS鑒定的方法可以很好地解決這個(gè)問(wèn)題。應(yīng)用該方法,低豐度的多肽和高豐度的多肽有同等的機(jī)會(huì)被鑒定出來(lái)。
美國(guó)西北太平洋國(guó)家實(shí)驗(yàn)室在2003年發(fā)表文章[29]稱將這種方法應(yīng)用于鑒定耐輻射奇球菌的蛋白質(zhì)組,使用的儀器是毛細(xì)管色譜串聯(lián)飛行時(shí)間質(zhì)譜(質(zhì)量精確度<10×10-6(10ppm))。首先,他們根據(jù)以前關(guān)于耐輻射奇球菌的液相色譜-串聯(lián)傅里葉變換離子回旋共振質(zhì)譜和LC-MS/MS實(shí)驗(yàn)的結(jié)果建立起包含多肽序列、精確質(zhì)量數(shù)和保留時(shí)間標(biāo)簽的數(shù)據(jù)庫(kù)。在這個(gè)數(shù)據(jù)庫(kù)中,多肽的保留時(shí)間被標(biāo)準(zhǔn)化到一個(gè)[0,1]區(qū)間里。接下來(lái),對(duì)新的LCMS實(shí)驗(yàn)譜圖中的每一個(gè)質(zhì)譜峰,如果可以在AM T tag數(shù)據(jù)庫(kù)中找到唯一的一個(gè)序列,使得它們的質(zhì)量數(shù)和保留時(shí)間的誤差不大于10×10-6和0.05單位時(shí)間,那么這個(gè)序列就與這個(gè)質(zhì)譜峰匹配。他們報(bào)道這種新的鑒定方式具有很高的靈敏度,但是并未考慮該方法的假陽(yáng)性率。在之后的幾年中,他們對(duì)這個(gè)算法作了一些改進(jìn)[30,31],如用多肽色譜峰的頂點(diǎn)代替其在質(zhì)譜中被檢測(cè)的時(shí)間,并應(yīng)用到多個(gè)不同的生物系統(tǒng)[32]和定量蛋白質(zhì)組的分析[33]中。該研究組于2007年開發(fā)出的VIPER軟件[34]可以自動(dòng)地進(jìn)行LC-MS譜圖中特征峰的檢測(cè)和比對(duì),從AM T tag數(shù)據(jù)庫(kù)中找到匹配的記錄,從而鑒定多肽序列和進(jìn)行定量分析。
另一類方法比AM T tag數(shù)據(jù)庫(kù)更具有一般性。這類方法不需要事先收集多肽序列和保留時(shí)間信息來(lái)建立數(shù)據(jù)庫(kù),而是只通過(guò)LC-MS比對(duì),直接尋找多次實(shí)驗(yàn)中精確質(zhì)量數(shù)和保留時(shí)間都十分接近的多肽特征峰,如果這些特征峰中有一部分已經(jīng)通過(guò)MS/MS鑒定出了序列,其他的峰也就隨之獲得了鑒定。PEPPeR[35]及SuperH irn[25]等都屬于這一類型的算法。
3.2 利用保留時(shí)間信息進(jìn)行M S/M S鑒定
在以往的研究中,數(shù)據(jù)庫(kù)搜索算法是從MS/MS譜圖鑒定多肽的主要方法。該算法的核心思想是將實(shí)驗(yàn)譜圖和數(shù)據(jù)庫(kù)中多肽的理論MS/MS譜圖進(jìn)行比對(duì),并對(duì)其匹配程度打分。但這種算法并不完美,其鑒定結(jié)果同時(shí)存在著假陽(yáng)性和假陰性的情況。保留時(shí)間作為一維新的參數(shù),可以幫助提高M(jìn)S/MS鑒定的準(zhǔn)確性和靈敏度。
一種方法是將保留時(shí)間參數(shù)和其他評(píng)價(jià)譜圖質(zhì)量的參數(shù)混合成一個(gè)新的參數(shù),這個(gè)新參數(shù)作為唯一的參數(shù),用來(lái)決策鑒定是否正確。Strittm atter等[36]提出了一個(gè)新的打分函數(shù),是5個(gè)參數(shù)的加權(quán)和,這5個(gè)參數(shù)分別是預(yù)測(cè)與實(shí)驗(yàn)保留時(shí)間誤差、質(zhì)量誤差和SEQU EST軟件產(chǎn)生的3個(gè)評(píng)價(jià)匹配質(zhì)量參數(shù)。參數(shù)的權(quán)重通過(guò)將一組由已知蛋白質(zhì)組成的混合物的MS/MS數(shù)據(jù)作為訓(xùn)練集進(jìn)行學(xué)習(xí)而獲得。應(yīng)用新的打分函數(shù),對(duì)該混合物的鑒定靈敏度增加了6.5%~9%(分別應(yīng)用果蠅、大鼠和人類的全蛋白質(zhì)組數(shù)據(jù)作為反相數(shù)據(jù)庫(kù)檢索)。另外,在鑒定人類血漿蛋白質(zhì)組時(shí),靈敏度增加了16%。
與此相反,另一些研究則把保留時(shí)間和譜圖匹配的打分參數(shù)分開使用。Kaw akam i等[10]把實(shí)驗(yàn)和預(yù)測(cè)的保留時(shí)間偏差當(dāng)作一個(gè)預(yù)篩選參數(shù),只有保留時(shí)間誤差在一定的容忍范圍內(nèi)時(shí)才能進(jìn)行隨后的多肽匹配步驟。
在Shen等[37]的方法中,當(dāng)譜圖的匹配質(zhì)量略低于較嚴(yán)格的標(biāo)準(zhǔn),但高于一個(gè)較寬松的標(biāo)準(zhǔn)時(shí),如果預(yù)測(cè)與實(shí)際保留時(shí)間的差別很小,同樣認(rèn)定這個(gè)MS/MS鑒定是正確的。Pfeifer等[38]也采用了類似的思想,他們用保留時(shí)間誤差作為過(guò)濾條件,從匹配的錯(cuò)誤概率大于1%但小于5%的MS/MS譜圖中篩選出高可信度的多肽鑒定。在不增加假陽(yáng)性率的前提下,使鑒定的多肽數(shù)目增加了19%。
除了預(yù)測(cè)的保留時(shí)間以外,經(jīng)驗(yàn)保留時(shí)間也被應(yīng)用于MS/MS鑒定。作者所在課題組[39]在2009年曾通過(guò)收集對(duì)同一樣品重復(fù)實(shí)驗(yàn)得到的高可信度的MS/MS鑒定和保留時(shí)間數(shù)據(jù),建立了經(jīng)驗(yàn)保留時(shí)間數(shù)據(jù)庫(kù),從匹配程度較差的MS/MS譜圖中篩選高可信度鑒定。雖然經(jīng)驗(yàn)數(shù)據(jù)庫(kù)因只收集了高可信度的經(jīng)MS/MS鑒定的多肽而使其包含的多肽數(shù)量有限,但避免了由于保留時(shí)間預(yù)測(cè)錯(cuò)誤造成的誤差,與預(yù)測(cè)的方法形成互補(bǔ)。
4 利用保留時(shí)間鑒定蛋白質(zhì)翻譯后修飾
蛋白質(zhì)的翻譯后修飾是蛋白質(zhì)組研究的重要課題。UN IMOD網(wǎng)站(http://www.un im od.org/)收錄的PTM已達(dá)數(shù)百種之多。傳統(tǒng)的檢測(cè)蛋白質(zhì)PTM的方法是對(duì)樣品進(jìn)行LC-MS/MS分析,然后采用數(shù)據(jù)庫(kù)搜索算法來(lái)鑒定一種或幾種已知的PTM的位點(diǎn)。考慮到同一個(gè)氨基酸殘基具有被修飾和未被修飾兩種質(zhì)量數(shù)不同的狀態(tài),而特定的PTM通常在幾個(gè)特殊的氨基酸殘基上出現(xiàn),鑒定PTM會(huì)造成數(shù)據(jù)庫(kù)檢索空間的數(shù)倍乃至數(shù)十倍的增加,在檢索時(shí)間增加的同時(shí),錯(cuò)誤匹配機(jī)率隨之上升。應(yīng)用數(shù)據(jù)庫(kù)檢索算法不可能實(shí)現(xiàn)同時(shí)檢索所有蛋白質(zhì)的PTM的任務(wù)。
在蛋白質(zhì)組樣品中,同一個(gè)多肽的翻譯后修飾和未被修飾的形式往往同時(shí)存在。基于這一現(xiàn)象,一些研究組通過(guò)對(duì)修飾和未修飾兩種形式的多肽的母離子m/z、碎片離子和保留時(shí)間的相關(guān)關(guān)系進(jìn)行PTM的鑒定。
Savitski等[40]發(fā)明的M odifiCom b算法利用高質(zhì)量精度的傅里葉變換質(zhì)譜數(shù)據(jù),可以不受限制地同時(shí)鑒定所有存在的PTM,即使是未知的PTM也能夠檢索到其質(zhì)量數(shù)。該算法主要利用MS/MS提供的多肽碎片離子信息。他們首先將用MASCO T軟件從MS/MS譜圖中鑒定到的高可信度的未被修飾的多肽序列作為研究的基礎(chǔ),若有另一張MS/MS譜圖與基準(zhǔn)多肽的MS/MS譜圖中有多個(gè)(通常定義為4個(gè))碎片離子的m/z相同,或者都相差某個(gè)固定的值,則認(rèn)為這個(gè)多肽很可能是基準(zhǔn)多肽被修飾之后的形式,它們的質(zhì)量差可以用來(lái)鑒定PTM的類型。他們發(fā)現(xiàn),同一PTM出現(xiàn)在不同的氨基酸殘基上會(huì)引起不同大小的保留時(shí)間偏移,因此應(yīng)用保留時(shí)間可以區(qū)分PTM的發(fā)生位點(diǎn)。
D asari等[41]根據(jù)質(zhì)量差和保留時(shí)間偏移的原理,在低分辨率的質(zhì)譜儀上鑒定了去酰胺化的多肽(天冬酰胺變?yōu)樘於彼?或谷氨酰胺變?yōu)楣劝彼?。由于質(zhì)量數(shù)相差只有0.984,傳統(tǒng)的數(shù)據(jù)庫(kù)檢索算法不能準(zhǔn)確地從低分辨率質(zhì)譜產(chǎn)生的數(shù)據(jù)中檢索到這一修飾。研究者通過(guò)人工合成的多肽,發(fā)現(xiàn)修飾和未修飾的多肽在強(qiáng)陽(yáng)離子交換色譜中的保留時(shí)間一致,而在反相色譜分離時(shí)去酰胺化的多肽晚3m in流出。利用保留時(shí)間差對(duì)MS/MS的鑒定結(jié)果進(jìn)行進(jìn)一步的判定,可以獲得93%的PTM鑒定準(zhǔn)確率,而通過(guò)人工視譜對(duì)MS/MS鑒定結(jié)果進(jìn)行判定的準(zhǔn)確率只有不足42%。
中國(guó)科學(xué)院計(jì)算技術(shù)研究所Fu等[42]于2009年發(fā)表論文,提出了一個(gè)高效的檢索樣品中高豐度蛋白質(zhì)的PTM的方法。和M odifiCom b一樣,該算法可以同時(shí)檢索所有可能存在的PTM。算法只計(jì)算多肽的母離子m/z和保留時(shí)間偏差,而不考慮MS/MS的信息,因此計(jì)算速度更快。首先計(jì)算所有譜圖兩兩間的質(zhì)量差,出現(xiàn)頻率很高且質(zhì)量差在0~100之間則作為可能的候選PTM進(jìn)入下一步的計(jì)算。由于修飾和未修飾的多肽只有一個(gè)修飾基團(tuán)的差別,其物理化學(xué)性質(zhì)比較接近,它們之間的保留時(shí)間差理論上是一個(gè)固定且較小的數(shù)值。基于這個(gè)假設(shè),可以利用二元(分別是質(zhì)量差和保留時(shí)間差)混合高斯模型來(lái)區(qū)分由PTM產(chǎn)生的或隨機(jī)產(chǎn)生的質(zhì)量差。在應(yīng)用于分析糖蛋白質(zhì)組的數(shù)據(jù)時(shí),該算法能夠比常規(guī)的數(shù)據(jù)檢索方法多解釋10%的譜圖。
現(xiàn)有的研究已經(jīng)表明,保留時(shí)間信息可以幫助研究者更加準(zhǔn)確、高效地進(jìn)行多肽的翻譯后修飾的鑒定。但是很少有研究涉及每種PTM給多肽的色譜行為帶來(lái)的確切影響[10,43],只假設(shè)PTM會(huì)產(chǎn)生保留時(shí)間的較小的恒定的偏移,對(duì)保留時(shí)間信息的利用還不夠充分。如果可以精確地為每種PTM預(yù)測(cè)可能產(chǎn)生的保留時(shí)間偏差,將能夠大大提高PTM鑒定的準(zhǔn)確度。
5 保留時(shí)間比對(duì)在定量蛋白質(zhì)組研究中的應(yīng)用
準(zhǔn)確可靠的定量蛋白質(zhì)組學(xué)研究方法是深入理解不同狀態(tài)生命的變化、為疾病診斷尋找生物學(xué)標(biāo)記的必要工具。LC-MS比對(duì)算法的發(fā)展使得多次實(shí)驗(yàn)間的非標(biāo)記定量成為可能,從而加快了生物標(biāo)志物發(fā)現(xiàn)的腳步。
其中一種定量方法是首先從LC-MS譜圖中提取多肽的色譜峰,再通過(guò)比較這些色譜峰的峰高或峰面積進(jìn)行定量。一個(gè)完整的定量算法一般包括以下幾個(gè)步驟:(1)MS譜圖的預(yù)處理;(2)信號(hào)的平滑與噪聲的去除;(3)特征峰的鑒定并計(jì)算其峰高和峰面積;(4)保留時(shí)間;(5)誤差評(píng)估;(6)特征峰分類,尋找生物標(biāo)記物。Radulovic等[44]開發(fā)的軟件平臺(tái)可以進(jìn)行以上所有步驟的自動(dòng)化處理。M etA lign軟件[45]是另外一個(gè)軟件,它最多可以對(duì)1 000次實(shí)驗(yàn)數(shù)據(jù)進(jìn)行比對(duì)。
提取多肽色譜峰進(jìn)行定量的方法有一個(gè)先天的不足,就是在提取色譜峰的過(guò)程中引入了額外的誤差,尤其是在處理低分辨率的數(shù)據(jù)時(shí),誤差更為明顯。為了避免這一問(wèn)題,Prakash等[46]直接分析未經(jīng)過(guò)預(yù)處理的二維LC-MS圖像(分別以時(shí)間和m/z為兩軸,信號(hào)強(qiáng)度顯示為點(diǎn)的顏色深淺的信號(hào)圖)。該算法首先根據(jù)名為CHAMS的D TW比對(duì)算法對(duì)原始的LC-MS圖像進(jìn)行比對(duì)。這個(gè)比對(duì)算法的特點(diǎn)是將峰強(qiáng)度列入考慮范圍,特征峰的提取步驟則放在比對(duì)之后。他們報(bào)道這種方法比先提取特征峰再比對(duì)的方法具有更高的特異性和靈敏度。
前面已經(jīng)對(duì)LC-MS比對(duì)算法進(jìn)行了詳細(xì)的介紹,此處不再贅述非標(biāo)記定量方法中與保留時(shí)間無(wú)關(guān)的其他技術(shù)環(huán)節(jié)。關(guān)于非標(biāo)記定量方法的介紹可見W ong等[47]和Am erica等[48]的綜述。
6 結(jié)論
本文介紹了多肽的保留時(shí)間信息在蛋白質(zhì)組數(shù)據(jù)分析的多個(gè)領(lǐng)域中的應(yīng)用情況。通過(guò)AM T tag數(shù)據(jù)庫(kù)或比對(duì)MS和MS/MS譜圖提高蛋白質(zhì)鑒定靈敏度的方法已經(jīng)得到了一定程度的應(yīng)用。利用保留時(shí)間也可以鑒定蛋白質(zhì)的翻譯后修飾。另外,在進(jìn)行非標(biāo)記定量時(shí),保留時(shí)間比對(duì)是必不可少的核心步驟。
遺憾的是,利用保留時(shí)間進(jìn)行MS/MS鑒定的研究仍處于算法的發(fā)明和驗(yàn)證階段,實(shí)際應(yīng)用很少。該方法的問(wèn)題在于,鑒定的準(zhǔn)確度強(qiáng)烈地依賴于保留時(shí)間預(yù)測(cè)方法的準(zhǔn)確度,而由于預(yù)測(cè)算法都是通過(guò)對(duì)有限大小的已知數(shù)據(jù)集進(jìn)行學(xué)習(xí)得到,其可推廣性很難得到保證。另外,由于對(duì)多肽的色譜行為的了解還不夠深入,利用保留時(shí)間進(jìn)行PTM鑒定也處于初級(jí)階段,因此保留時(shí)間目前只作為鑒定的輔助信息。盡管存在著一些問(wèn)題,現(xiàn)有的研究已經(jīng)證明了保留時(shí)間作為獨(dú)立于質(zhì)譜數(shù)據(jù)的參數(shù),可以提高蛋白質(zhì)鑒定和定量的準(zhǔn)確度及效率。隨著實(shí)驗(yàn)數(shù)據(jù)的積累和對(duì)多肽色譜行為更深入的研究,保留時(shí)間將在今后的蛋白質(zhì)組研究中發(fā)揮更重要的作用。
[1] Guo D,M ant C T,Taneja A K,et al.J Chromatogr,1986,359:499
[2] Mant C T,Burke T W L,B lack J A,et al.J Chromatogr,1988,458:193
[3] Browne C A,Bennett H P J,Solom on S.Anal B iochem,1982,124:201
[4] Meek J L.Proc Natl Acad SciUSA,1980,77:1632
[5] Meek J L,Rossetti Z L.J Chromatogr,1981,211:15
[6] Sakamoto Y,Kaw akam i N,Sasagawa T.J Chromatogr,1988,442:69
[7] Petritis K,Kangas L J,Ferguson P L,et al.Anal Chem,2003,75(5):1039
[8] Guo D,M ant C T,Taneja A K,et al.J Chromatogr,1986,359:518
[9] Krokhin O V,Craig R,Spicer V,et al.mol Cell Proteomics,2004,3(9):908
[10] Kawakami T,Tateishi K,Yam ano Y,et al.Proteomics,2005,5(4):856
[11] Petritis K,Kangas L J,Yan B,et al.Anal Chem,2006,78(14):5026
[12] Baczek T,W iczling P,M arszallM,et al.J Proteome Res,2005,4(2):555
[13] Kaliszan R,Baczek T,Cim ochow ska A,et al.Proteomics,2005,5(2):409
[14] Salgado J C,Rapaport I,Asenjo J A.J Chromatogr A,2005,1098:44
[15] Salgado J C,Rapaport I,Asenjo J A.J Chromatogr A,2005,1075:133
[16] Salgado J C,Rapaport I,Asenjo J A.J Chromatogr A,2006,1107:120
[17] Salgado J C,Rapaport I,Asenjo J A.J Chromatogr A,2006,1107:110
[18] Old W M,Meyer-Arendt K,Aveline-Wolf L,et al.mol Cell Proteomics,2005,4(10):1487
[19] Li X J,Yi E C,Kemp C J,et al.Mol Cell Proteomics,2005,4(9):1328
[20] VandenbogaertM,Li-Thiao-Te S,Kaltenbach H M,et al.Proteomics,2008,8(4):650
[21] Bylund D,Danielsson R,Malmquist G,et al.J Chromatogr A,2002,961(2):237
[22] Christin C,Sm ilde A K,Hoefsloot H C,et al.Anal Chem,2008,80(18):7012
[23] Eilers P H.Anal Chem,2004,76(2):404
[24] Listgarten J,Neal R M,Roweis S T,et al.Bioinformatics,2007,23(2):e198
[25] M ueller L N,Rinner O,Schmidt A,et al.Proteomics,2007,7(19):3470
[26] Fischer B,Grossm ann J,Roth V,et al.B ioinformatics,2006,22(14):e132
[27] Fischer B,Roth V,Buhmann J M.BMC B ioinformatics,2007,8(Supp l10):S4
[28] Podwojski K,Fritsch A,Cham rad D C,et al.Bioinformatics,2009,25(6):758
[29] Strittmatter E F,Ferguson PL,Tang K,et al.J Am Soc mass Spectrom,2003,14(9):980
[30] Zimm er J S,M onroe M E,Q ian W J,et al.mass Spectrom Rev,2006,25(3):450
[31] Kiebel G R,Auberry K J,Jaitly N,et al.Proteom ics,2006,6(6):1783
[32] Adkins J N,Monroe M E,Auberry K J,et al.Proteom ics,2005,5(13):3454
[33] M anes N P,Estep R D,Mottaz H M,et al.J Proteom e Res,2008,7(3):960
[34] M onroe ME,Tolic N,Jaitly N,et al.B ioinform atics,2007,23(15):2021
[35] Jaffe J D,M ani D R,Lep tos K C,et al.Mol Cell Proteom ics,2006,5(10):1927
[36] Strittm atter E F,Kangas L J,Petritis K,et al.J Proteom e Res,2004,3(4):760
[37] Shen Y,Kim J,Strittm atter E F,et al.Proteom ics,2005,5(15):4034
[38] Pfeifer N,Leinenbach A,Huber C G,et al.J Proteom e Res,2009,8(8):4109
[39] Sun W,Zhang L,Yang R,et al.Rap id Comm un Mass Spectrom,2009,23(1):109
[40] Savitski M M,N ielsen M L,Zubarev R A.mol Cell Proteom ics,2006,5(5):935
[41] Dasari S,W ilm arth P A,Rustvold D L,et al.J Proteom e Res,2007,6(9):3819
[42] Fu Y,J ia W,Lu Z,et al.BMC B ioinform atics,2009,10(Supp l1):S50
[43] Kim J,Petritis K,Shen Y,et al.J Chromatogr A,2007,1172(1):9
[44] Radulovic D,Jelveh S,Ryu S,et al.mol Cell Proteom ics,2004,3(10):984
[45] Lomm en A.Anal Chem,2009,81(8):3079
[46] Prakash A,M allick P,Whiteaker J,et al.mol Cell Proteom ics,2006,5(3):423
[47] W ong J W,Sullivan M J,Cagney G.B rief B ioinform,2008,9(2):156
[48] Am erica A H,Cordew ener J H.Proteom ics,2008,8(4):731
Application of peptide retention time in proteome research
SHAO Chen*,GAO Youhe
(Department of Physiology and Pathophysiology,Institute of Basic Medical Sciences,Chinese Academy of Medical Sciences,School of Basic Medicine,Peking Union Medical College,Beijing 100005,China)
O658
A
1000-8713(2010)02-0128-07
*通訊聯(lián)系人:邵 晨,助理研究員,主要研究方向?yàn)榈鞍踪|(zhì)組學(xué)和生物信息學(xué).Tel:(010)65296407,E-m ail:scshaochen@126.com.
中國(guó)醫(yī)學(xué)科學(xué)院基礎(chǔ)醫(yī)學(xué)研究所院(所)長(zhǎng)基金項(xiàng)目(No.2009PY05)、國(guó)家自然科學(xué)基金杰出青年基金項(xiàng)目(No.30725009)、國(guó)家自然科學(xué)基金項(xiàng)目(No.30870502)、北京市自然科學(xué)基金項(xiàng)目(No.5072037)和高等學(xué)校博士學(xué)科點(diǎn)專項(xiàng)科研基金項(xiàng)目(No.20070023021).
2009-12-24
DO I:10.3724/SP.J.1123.2010.00128
猜你喜歡
小獼猴智力畫刊(2022年9期)2022-11-04 02:31:42
中學(xué)生數(shù)理化·中考版(2022年11期)2022-02-16 07:01:20
小哥白尼(趣味科學(xué))(2019年6期)2019-10-10 01:01:50
兒童故事畫報(bào)(2019年5期)2019-05-26 14:26:14
意林原創(chuàng)版(2016年10期)2016-11-25 10:28:30
發(fā)明與創(chuàng)新(2016年38期)2016-08-22 03:02:52
太空探索(2016年5期)2016-07-12 15:17:55
Coco薇(2016年2期)2016-03-22 02:42:52
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長(zhǎng)指南(2015年7期)2015-08-11 15:03:12
