DIR-MCFC中催化劑抗堿中毒的研究
2010-09-18 06:20:30張雄福
電池 2010年5期
關鍵詞:催化劑
涂 敏,張 健,張雄福,周 利
(1.大連理工大學化工學院,精細化工國家重點實驗室,遼寧大連 116012;2.中國科學院大連化學物理研究所,潔凈能源國家實驗室,遼寧大連 116023)
熔融碳酸鹽燃料電池(MCFC)的電解質通常為Li2CO3和K2CO3的混合物[n(Li2CO3)∶n(K2CO3)=62∶38]。在高溫(650℃)下,電解質中的能自由流動,在電場的作用下從陰極流向陽極,提供離子傳導。在陽極,H2與遷移來的在電極三相界面反應,生成CO2和H2O,并釋放電子;陽極產生的電子由外電路來到陰極,與O2、CO2反應,生成。MCFC在建立高效、環境友好的50~1 000 kW固定式電源或分散式電站方面有優勢,可減少CO2排放40%以上,并實現熱-電聯供或聯合循環發電,將燃料利用率提高到70%~80%[1]。我國對MCFC的研究起步較晚,雖已在2001年進行了1 kW的MCFC發電實驗[2-3],但與歐美、日本相比,還有差距。
可將重整制氫反應設備置于MCFC內,構成直接內重整MCFC(DIR-MCFC)。DIR-MCFC作為整體技術的優勢在于[4]:不需外部重整器,降低了成本;可同時實現電化學、重整反應的物質和能量耦合;電極反應陽極消耗氫可打破重整反應熱力學平衡,提高甲烷轉化率和發電效率,系統總效率可提高近12%;陽極的氫分布更均勻,溫度分布更理想。
由于電解質Li2CO3和K2CO3在熔融狀態下依靠毛細作用吸附于多孔電解質隔膜中,熔融電解質會隨著DIR-MCFC的運行而流失,導致隔膜大孔不能完全充滿融鹽電解質,燃料和氧化劑發生互串,最終使電池失效。流失的熔融電解質(氣態擴散或液態蠕爬)還會引起催化劑的失活,導致電池效率降低乃至失效,阻礙了DIR-MCFC的商業化。提高催化劑抗堿中毒能力、解決堿污染問題,是延長DIR-MCFC運行壽命、降低成本及維持系統效率的主要舉措之一[5]。
1 內重整催化劑及堿中毒問題
在DIR-MCFC中,除了電解質的腐蝕、陰極的溶解、陽極蠕變、隔膜燒結等系統關鍵問題需進一步的研究和改進外,還存在催化劑堿中毒的問題。由于催化劑置于陽極室內氣體分布孔道中,隨著電池的運轉,電解質隔膜中的熔融碳酸鹽便會以氣態或液態的形式發生擴散或蠕爬作用而接觸到催化劑,痕量的堿蒸氣或堿液都會引起重整催化劑失活[2]。
M.Matsumura等[6]發現,電解質通過氣態擴散的方式接觸到催化劑是催化劑中毒的主要原因,而液態堿的蠕爬起次要作用。在電解液的氣態擴散中,K、Li的擴散速度分別為4.4×10-11mol/(cm2·s)和 1.7×10-12mol/(cm2·s),由于擴散速度差異大,在收集過程中,K能很短時間內達到飽和,飽和度約為1.1%;而Li隨著收集時間的延長,含量逐漸提高。
痕量堿使內重整催化劑失活的相關機理,人們有不同的看法。通過量子力學計算Ni(111面),發現吸附的K對Ni原子表面的影響很大,利用有效媒質理論的靜電學計算,證明堿的存在能促進Ni催化劑對蒸汽的吸附,但堿的增加不能改變甲烷的吸附能和活化能。由于甲烷的非極性,該理論不適用于甲烷的活化過程,但可用于乙醇等的活化過程[7]。
J.R.Rostrup-Nielsen等[8]對7 kW DIR-MCFC電堆進行中試實驗,條件為 3 500 h、n(H2O)∶n(C)=4∶1、P=0.1 M Pa、n(Li2CO3)∶n(K2CO3)=62∶38。 將 Ni-MgAl2O4系列催化劑分別填在冷卻通道和陽極室中,發現電池運行后馬上觀測到緩慢的堿中毒現象,反應后陽極室中催化劑表面K+含量達1.6%~4.5%,甲烷平衡轉化率下降20%。研究發現,堿造成Ni基或貴金屬催化劑的內在活性嚴重降低,其中K起主要作用,而Li次之。堿的存在并未增強鎳晶體的燒結作用,有些載體燒結是由于堿促進了催化劑對蒸汽的吸附,隨著蒸汽量的增加促使催化劑燒結。催化劑的失活,可能是因為Ni晶粒的高活性面被刻蝕成低活性面(111面)。
在同樣堿蒸汽分壓下,酸性載體如Al2O3等能比一般性載體如 MgO,吸附更多的堿,更易發生中毒。S.Cavallaro等[9]對MgO載體的催化劑研究發現,在DIR-MCFC測試條件下可維持860 h,催化劑失活是孔道堵塞。原始催化劑在測試前無 K和Li,測試后含有 0.59%~1.40%Li和5.2%~8.7%K。催化劑小球的整個外孔道被KOH玻璃層堵塞,阻止了反應物到達催化劑的活性位,影響了甲烷重整反應[3,9]。
R.J.Berger等[10]用含濕浸潤法在α-Al2O3和γ-Al2O3載體上合成Ni系重整催化劑,用于電池外固定床重整檢測,條件為650℃、n(Li2CO3)∶n(K2CO3)=62∶38,反應100 h后,催化劑表面 K(KOH)、Li(Li2O)的含量分別達0.35%和2.3%,重整反應速率常數由4×10-4m3/(gcal·s)降至 0.5×10-4~ 0.6×10-4m3/(gcal·s),Ni晶粒由 14~ 47 nm 增至100 nm以上。催化劑失活的原因有:①Ni晶粒由于堿的存在長至50~100 nm或更大;②由于對堿的吸附,大部分的Ni活性位失活;③堿與Ni或其他吸附物質發生界面間化學反應;④載體物質的燒結;⑤堿與含Al2O3載體發生化學反應;⑥堿覆蓋了催化劑大部分的內表面,進而堵塞催化劑孔道。
K.Sugiura等[11]運用非接觸攝像技術(高空間分辨率攝像機),在電池面積為16 m2的單體MCFC中進行原位揮發性物質觀測,條件為650℃、n(Li2CO3)∶n(K2CO3)=62∶38,發現:隨著電流密度的增加,從陽極通道入口到出口出現的、由揮發性KOH、LiOH組成的氣狀光帶的亮度和濃度都在增大。KOH由電解質中的K2CO3與電池中生成的H2O反應得到,揮發出的KOH接觸催化劑后,與重整產物CO2生成K2CO3并沉積在催化劑表面,導致催化劑中毒。
L.Ho-In等[12]對MgO為載體的Ni催化劑抗堿中毒的研究發現,當20%Li與催化劑混合后,制備的Ni/MgO完全失活。通過程序升溫還原(TPR)、XRD分析等發現:堿金屬化合物由晶態變為玻璃態,不僅導致催化劑的物理堵塞,而且Li與載體生成 LiyNixMg1-x-yO固溶體,覆蓋了Ni活性位,使催化劑失活。在載體中加入TiO2,可解決堿中毒問題。
DIR-MCFC催化劑堿中毒機理和積碳機理的研究很多,通常認為:堿和碳沉積,覆蓋在催化劑表面,堵塞催化劑內部孔道,誘發了催化劑的燒結,侵占了重整反應活性位或刻蝕活性面,使甲烷轉化率下降,電池發電效率降低。這些研究對DIR-MCFC催化劑阻堿工作有很好的指導作用,但在堿蒸汽和堿液的物化性質(分子態、擴散性、潤濕性、反應活性等)、污染催化劑能力(堿中毒主導作用)、堿中毒催化劑再生等方面,有待進一步的研究。
2 抗堿中毒研究進展
目前,針對以消除堿擴散和防止重整催化劑中毒為目的研究,可歸結為以下3種方案[3]。
2.1 催化劑改良
人們最初通過添加Ru、Rh等貴金屬或改良催化劑載體(MgO、ZrO2、γ-Al2O3、LiAlO2)對催化劑進行抗堿改性。
K.Harima等[13]制備的Ru/ZrO2內重整催化劑在運行2 700 h后,活性降低不明顯,具有很好的抗堿中毒效果;但貴金屬價格高昂,不利于大規模使用。
起初,研究人員主要采用在抗堿性載體(如MgO、LiAlO2等)上負載Ni,制備抗堿中毒內重整催化劑,并取得了較好的阻堿效果[9]。R.J.Berger等[10]指出,這些研究中催化劑中毒時間太短,且測試條件沒有電池實際運行時苛刻。
若測試條件比電池實際運行時苛刻,采用抗堿性載體的催化劑阻堿效果較差,可在載體中加入一些物質,防止催化劑中毒。L.Ho-In等[12-13]采用浸漬沉積法制備了一系列Ni/MgO-TiO2重整催化劑,并在石英管固定床反應器中[650℃、n(H2O)∶n(C)=2.5∶1.0]進行池外抗堿實驗,Li2CO3與催化劑物理混合,其中Li2CO3的含量為0~20%,發現富Ti和貧Ti系列催化劑在接觸Li后迅速失活,而n(Mg)∶n(Ti)=2∶1~7∶1時,甲烷轉化率保持在無堿時的 60%。TPR、XRD分析認為,在催化劑中引入TiO2,生成 Mg-Ti混合氧化物,阻止了NixMg1-xO固溶體的形成,增強了抗堿中毒能力,且當n(Mg)∶n(Ti)=2∶1時效果最好。
K.Park等[14]通過雙層氫氧化物(LDH)前驅體,制備了系列Ni-Mg-Al重整催化劑[n(Al)∶n(Mg)=0.11∶1.00~2.00∶1.00],并在石英管固定床反應器中[650℃、n(H2O)∶n(C)=2.5∶1.0]進行池外抗堿實驗(物理混合 Li2CO3、10%~40%),發現制備的催化劑在不與堿混合時,轉化率比傳統的商業催化劑要高。采用富鋁催化劑[n(Al)∶n(Mg)>1∶1]時,與堿混合后甲烷的轉化率能達到80%以上,具有很好的抗堿中毒效果。XRD分析認為,催化劑中的Al2O3與Li反應后生成γ-LiAlO2,保護了Ni活性位不被Li覆蓋。比較發現,n(Al)∶n(Mg)=1.5∶1.0的催化劑最為理想,比表面積最大,Ni晶粒最小,催化活性最高,抗堿中毒最佳。
改良催化劑載體,可增加催化劑的抗堿中毒效果,但作用有限,且堿蒸氣或堿液對多孔載體都有吸附、堵塞、腐蝕作用。雖然如此,研究高抗積碳重整催化劑仍有重要的意義。
周敬林[15]設計了Silicalite-1沸石膜包覆型核-殼重整催化劑,采用沸石生長法,分別在Ni-SiO2和Ni-Al2O3催化劑表面生長一層連續的Silicalite-1沸石膜,形成核-殼催化劑。利用多孔沸石殼層對堿性毒害物質的阻隔或吸附作用,將堿性物質阻截或滯留于殼層而不直接接觸催化活性中心;催化劑核可維持催化活性使反應持續供氫,H2O、CH4、H2、CO及CO2分子可通過擴散順利透過Silicalite-1膜層。不同催化劑的甲烷轉化率[650 ℃、n(H2O)∶n(C)=3∶1]見圖1[15]。
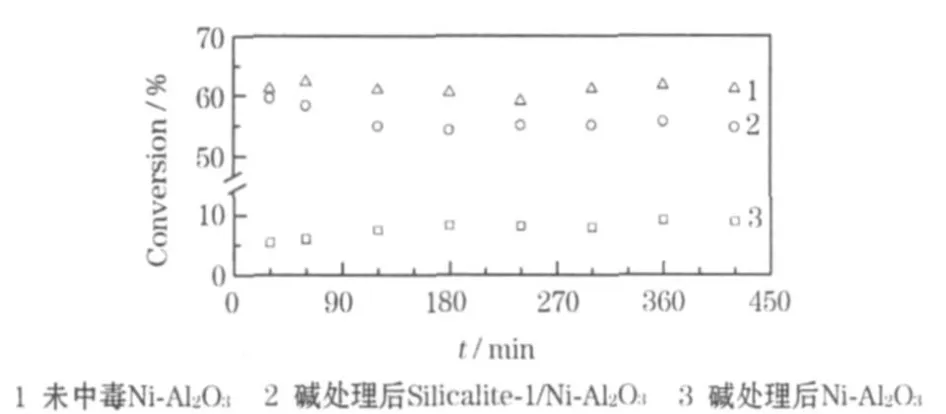
圖1 堿處理后催化劑SRM反應甲烷轉化率隨時間的變化Fig.1 The changes of methane conversion of different catalysts SRM reaction with time
通過池外測試發現,在Ni-Al2O3原始催化劑上甲烷轉化率始終保持在62%左右,堿中毒處理后,Ni-Al2O3催化劑的甲烷轉化率只有8%左右,甲烷轉化率大輻降低;在Ni-Al2O3表面包覆Silicalite-1沸石膜層后形成核-殼催化劑,堿處理后的甲烷轉化率能保持在56%左右,與Ni-Al2O3原始催化劑相近,遠高于堿處理后的Ni-Al2O3催化劑。這表明制備的核-殼催化劑具有較強的抗堿中毒能力。
2.2 設置阻堿隔板或保護箔
在催化劑與陽極室之間設置惰性保護屏或防護板,可延長催化劑的壽命。保護屏可作為化學阱或物理阱,阻隔、排斥堿氣而自由通過氫和水分子,保護催化劑和維持反應。
L.G.Marianowski[16]采用雙室陽極結構,在燃料室和陽極室之間安置一層厚度為0.25~2.50 mm的致密金屬箔(Cu、Ni),將催化劑填于燃料室中,催化產生的氫分子在金屬箔上解離滲透,進入陽極室反應,而堿性電解質、氫分子等不能透過金屬箔,保護了重整催化劑,延長了電池的壽命。
Y.Miyake等[17]在陽極氣體通道內置入一層鍍鎳隔板,該隔板將Ni鍍在波浪形的鉻鎳鐵合金集流板上,可阻止液態堿的蠕爬,延長DIR-MCFC的壽命。
E.Passalacqua等[18]采用流延法制備約0.36 mm厚、平均孔徑為2μ m、孔隙率為 39%~43%的系列SiC陶瓷膜,并分別進行了 150 h、700 h和 1 500 h的池外測試,發現測試150 h后,SiC陶瓷膜可阻止50%~65%的K,但隨著測試時間延長到1 500 h,只有32%的K被截留在陶瓷膜上。
張雄福等[19]將電池的集流板設計成孔徑為0.35 mm的小孔集流板,并采用固態粒子燒結法,在電池的集流板穿孔孔壁上覆蓋一層致密非極性陶瓷膜。
通過構建致密光滑的陶瓷膜層,根據熔融碳酸鹽和陶瓷膜的極性差異,集流板的孔道可利用毛細現象來阻隔液態熔融碳酸鹽滲透過集流板,在防止熔融碳酸鹽流失的同時避免其與內重整催化劑接觸,防止催化劑的堿中毒,從而延長電池的使用壽命。這種集流板的設計不影響集流板的導電、導熱、透氣性能和機械強度,也不改變電池的原有結構。目前,涉及阻堿隔板的空間安置工藝、運行時間、機械強度、孔徑及厚度、阻堿機理方面等,還有待進一步研究。
2.3 優化操作條件
可通過優化操作條件,如改變陽極氣室內擔載催化劑的位置、n(H2O)∶n(C)和保持均勻的氣體分布等,來降低堿中毒。K.Sugiura等[20]認為,內重整催化劑中毒主要是由于KOH和K2CO3粘附在催化劑上,只要將其除掉就能使催化劑再生。根據反應,KOH能夠在惰性氣氛下揮發,但K2CO3不能,因此改變氣體組成,將K2CO3轉變為KOH并將其除掉,就能將中毒催化劑再生。催化劑在100%N2的氣氛中時,中毒催化劑的活性能恢復到中毒前的80%。當CO2濃度大于25%時,由于大部分KOH轉變為K2CO3,中毒催化劑很難再生。當燃料中的n(H2O)∶n(C)>2∶1時,DIRMCFC中催化劑基本上不會中毒。此外,將催化劑置于陽極的上端,使H2O含量增加,而CO2含量減少,有助于避免催化劑中毒。J.H.Wee[21]設計了一種梯形內重整器。該重整器運行200 h后,每個電池單元的抗電阻性均維持在使用傳統重整器的94%左右,而K和Li的中毒程度只有傳統重整器的54%和45%。主要原因是梯形催化床中不同的位置催化劑的載量不同,使整個重整器的溫度分布趨于均一,且陽極腔出口溫度比傳統重整器低8℃,低溫雖然增加了積炭,但減少了電解質的揮發,減輕了催化劑的中毒程度。
優化重整催化劑空間分布密度,以平衡重整反應吸熱和電化學放熱效應,是近年來的研究熱點。重整氣體入口的催化劑密度低,出口密度高,可抑制重整反應冷點、熱點的出現,平衡溫度分布,控制堿氣揮發量和擴散速度,在一定程度上抑制堿中毒,有助于提高電池系統效率和電效率[22]。
優化操作條件可在一定程度上減少堿中毒,但不能從根本上解決堿中毒的問題,隨著電池運行時間的延長,催化劑仍會逐漸失活。在以上3種方案中,制備抗堿中毒催化劑和設置阻堿隔板,是從根本上解決堿擴散和催化劑中毒的手段。目前,上述抗堿催化劑和阻堿隔板放入,池外測試都取得了一定的進展,但也僅限于池外實驗,還未與MCFC組配進行聯試。優化操作條件或改變電池結構,是電池設計和操作過程中正常考慮的問題,直接內重整催化劑的擔載位置和均勻的氣體分布流場設計,都是這種集成技術發展過程中最基本的出發點和組成部分。提高n(H2O)∶n(C),可在一定程度上減緩催化劑的堿中毒,但代價是降低電池的性能。
3 結語
內重整催化劑堿中毒問題一直是DIR-MCFC的一個重要的問題,要解決,還要從以下幾個方面進行全面考慮:
抗堿中毒的內重整催化劑還處于實驗室測試階段,阻礙應用的主要原因是催化劑壽命不能滿足電池長時間運行的要求。開發耐堿或具有阻堿功能,低成本、高效率、長壽命的內重整催化劑,是DIR-MCFC商業化的一個重要前提。
阻堿隔板可防止催化劑與電解質接觸,但很多設計導致電池結構復雜化,降低了電池性能和效率。阻堿隔板不但要解決電解質流失的問題,還要求長壽命,不影響電池的結構和效率,并與集流板有機整合,讓H2、CO、H2O等順利通過而阻止堿性電解質通過。此外,提高集流板在高溫下的電導率,也是提高電池效率的一種方法。
對于改進DIR-MCFC裝置的設計及操作條件,可將實驗與數學模擬綜合起來研究,通過模擬電池運行的熱、電性能來指導電池的設計。
[1]XIAO Gang(肖鋼).燃料電池技術[M].Beijing(北京):Publishing House of Electronics Industry(電子工業出版社),2009.
[2]YU Li-jun(于立軍),YUAN Jun-qi(袁俊琪),CAO Guang-yi(曹廣益).千瓦級熔融碳酸鹽燃料電池的實驗研究[J].Journal of Shanghai Jiao Tong University(上海交通大學學報),2001,37(9):1 391-1 401.
[3]ZHOU Li(周利),YI Bao-lian(衣寶廉),CHENG Mo-jie(程謨杰),et al.千瓦級熔融碳酸鹽燃料電池組[J].Battery Bimonthly(電池),2002,32(3):138-141.
[4]Dicks A L.Advances in catalysts for internal reforming in high temperature fuel cells[J].J Power Sources,1998,71(1-2):111-122.
[5]Koji N.An overview of the fuel cell and hydrogen technology development policies in Japan[J].J Chem Eng Jpn,2006,39(5):489-502.
[6]Matsumura M,Hirai C.Transport mechanism of electrolyte vapor to reforming catalyst[J].Ind Eng Chem Res,1998,37(5):1 793-1 798.
[7]Simon D,Bigot B.A theoretical study of the electronic properties of a nickel(111)surface partially covered by potassium adatoms[J].Surf Sci,1994,306(3):459-470.
[8]Rostrup-Nielsen J R,Christiansen L J.Internal steam reforming in fuel cells and alkali poisoning[J].Appl Catal,A:Gen,1995,126(2):381-390.
[9]Cavallaro S,Freni S,Cannistraci R,et al.Alkali effect on the MCFC-internal reforming catalyst life[J].Int J Hydrogen Energy,1992,17(3):181-186.
[10]Berger R J,Doesburg E B M,van Ommen J G,et al.Nickel catalysts for internal reforming in molten carbonate fuel cells[J].Appl Catal,A:Gen,1996,143(2):343-365.
[11]Sugiura K,Yodo T,Yamauchi M,et al.Visualization of electrolyte volatile phenomenon in DIR-MCFC[J].J Power Sources,2006,157(2):739-744.
[12]Ho-In L,Choi J S,Jung-Sook Y,et al.Effect of lithium carbonate on nickel catalysts for direct internal reforming MCFC[J].J Power Sources,2005,145(2):652-628.
[13]Harima K,Yasuo M,Saito T,et al.Internal reforming molten carbonate fuel cells[P].JP:JP05335031,1992-05-29.
[14]Park K,Kim K Y,Lu L,et al.Structural characteristics of(Ni-MgAl)Oxprepared from a layered double hydroxide precursor and its application in direct internal reforming molten carbonate fuel cells[J].Fuel Cells,2007,7(3):211-217.
[15]ZHOU Jing-lin(周敬林).Silicalite-1沸石膜的合成及其核-殼催化劑的設計與應用[D].Dalian(大連):Dalian University of Technology(大連理工大學),2009.
[16]Marianowski L G.Dual compartment anode structure[P].US:4702973,1987-10-27.
[17]Miyake Y,Nakanishi N,Nakajima T,et al.A study on degradation phenomena of reforming catalyst in DIR-MCFC[J].J Chem Eng Jpn,1995,21(6):1 104-1 109.
[18]Passalacqua E,Freni S,Barone F.Alkali resistance of tape-cast SiC porous ceramic membranes[J].Mater Lett,1998,34(3-6):257-262.
[19]ZHANG Xiong-fu(張雄福),ZHANG Jian(張健),T U Min(涂敏),et al.一種復合阻堿集流板的制備方法[P].CN:201010301260.4,2009-02-05.
[20]Sugiura K,Daimon M,Tanimoto K.Optimum operating conditions of DIR-MCFC without vapor-phase carbonate pollution[J].J Power Sources,2003,118(1-2):228-236.
[21]Wee J H.Performance of a unit cell equipped with a modified catalytic reformer in direct internal reforming-molten carbonate fuel cell[J].J Power Sources,2006,156(2):288-293.
[22]Heidebrecht P,Sundmacher K.Optimization of reforming catalyst distribution in a cross-flow molten carbonate fuel cell with direct internal reforming[J].Ind Eng Chem Res,2005,44(10):3 522-3 528.
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
