X-染色體連鎖智力障礙相關基因研究進展
2010-09-10 01:38:38張文琴楊璐綜述馬端審校
中國產前診斷雜志(電子版) 2010年3期
張文琴 楊璐 綜述 馬端 審校
(1.上海市閔行區中心醫院,上海 201100;2.復旦大學出生缺陷研究中心,復旦大學分子醫學教育部重點實驗室,復旦大學生物醫學研究院,上海 200032)
先天性智力障礙是由中樞神經系統(CNS)發育異常引起的復雜性疾病,患者通常在18歲以前出現智力和行為方面的明顯缺陷。據統計,大約1%~3%的人存在智力障礙,男女比例大約為1.4~1.6:1[1]。在所有的智力障礙患者中,大約25%~35%與遺傳有關。在中重度智力障礙者中,遺傳因素約占50%。引起先天性智力障礙的因素包括染色體非整倍體、染色體結構異常、基因組疾病和單基因疾病[2]。
X-染色體連鎖智力障礙(X-linked mental retardation,XLMR)是一類由X染色體上基因突變引起的智力障礙,目前已經發現200余種,約占所有先天性智力障礙的25%[2]。在這些XLMR中,已確定149種為綜合征型(syndromic XLMR,MRXS),66種為非綜合征型(non-syndromic XLMR,MRX)[3,4]。XLMR發生率在男性約1/600~1/1 000[2]。
與常染色體遺傳病相比,X-連鎖遺傳病有3個特點:①男性X染色體上的基因為半合子,因此不論致病基因為顯性或隱性,都可導致男性發病;②男性患者的X-連鎖基因只能來自母親并只能傳給女兒,不存在“父-子”傳遞現象;③女性雜合子攜帶者是否有臨床表現不僅取決于致病基因的表達狀況,而且與X染色體是否失活有關。即有些女性雜合子有臨床表現,有些則沒有。
1 X染色體的概況
2005年3月17日,《Nature》雜志刊登了英國Ross MT領導的國際科學家小組對X染色體的測序結果,為X染色體的研究添加了最富有成效的內容。他們發現,X染色體有1.5億對堿基,容納1 100個基因,約占人類基因組所有基因的5%[5]。正如Ross所說:“從遺傳模式、獨一無二的生理特性以及與人類疾病等方面來看,X染色體絕對是人類基因組中最不同尋常的。”
2 XLMR相關基因
迄今為止,已發現90個XLMR相關基因,它們的 基 本 情 況 見 表 1(http://xlmr.interfree.it/home.htm)。
3 幾種新發現的XLMR基因
3.1 甲基Cp G結合蛋白2基因(MECP2)MECP2基因位于Xq28,是甲基化-Cp G結合蛋白家族中成員,其功能為部分基因的轉錄抑制劑。大約80%的Rett綜合征患者有 MECP2基因突變[6,7]。
Rett綜合征是一種進行性神經系統疾病,主要影響女性(發生率為1/10 000)。經過6~18個月的正常發育后,受累兒童開始失去一些獲得性能力,如手和語言的使用,同時出現頭圍增長緩慢,社會行為逐漸削弱,許多女孩發展為癲癇和焦慮,絕大多數患兒出現連續性手的刻板動作[8]。
3.2 肌酸轉運蛋白基因(SLC6A8) SLC6A8位于Xq 28,突變后可導致男性重度智障,而在大約半數
的女性攜帶者中,則表現為輕度的學習障礙[9]。患者的特征性表現是身材矮小、低體重、肌肉發育不良、肌張力減退、運動障礙、癲癇、行為和語言表達困難。通過質子磁力共振光譜,發現患者大腦中肌酸和磷酸肌酸完全缺乏,肌酸和肌酐在血漿和尿液中的比例增加,但是胍基乙酸水平正常,據此可以對該病進行臨床診斷。對于肌酐比率異常或肌酸缺乏的患者,應對SLC6A8基因進行DNA分析。
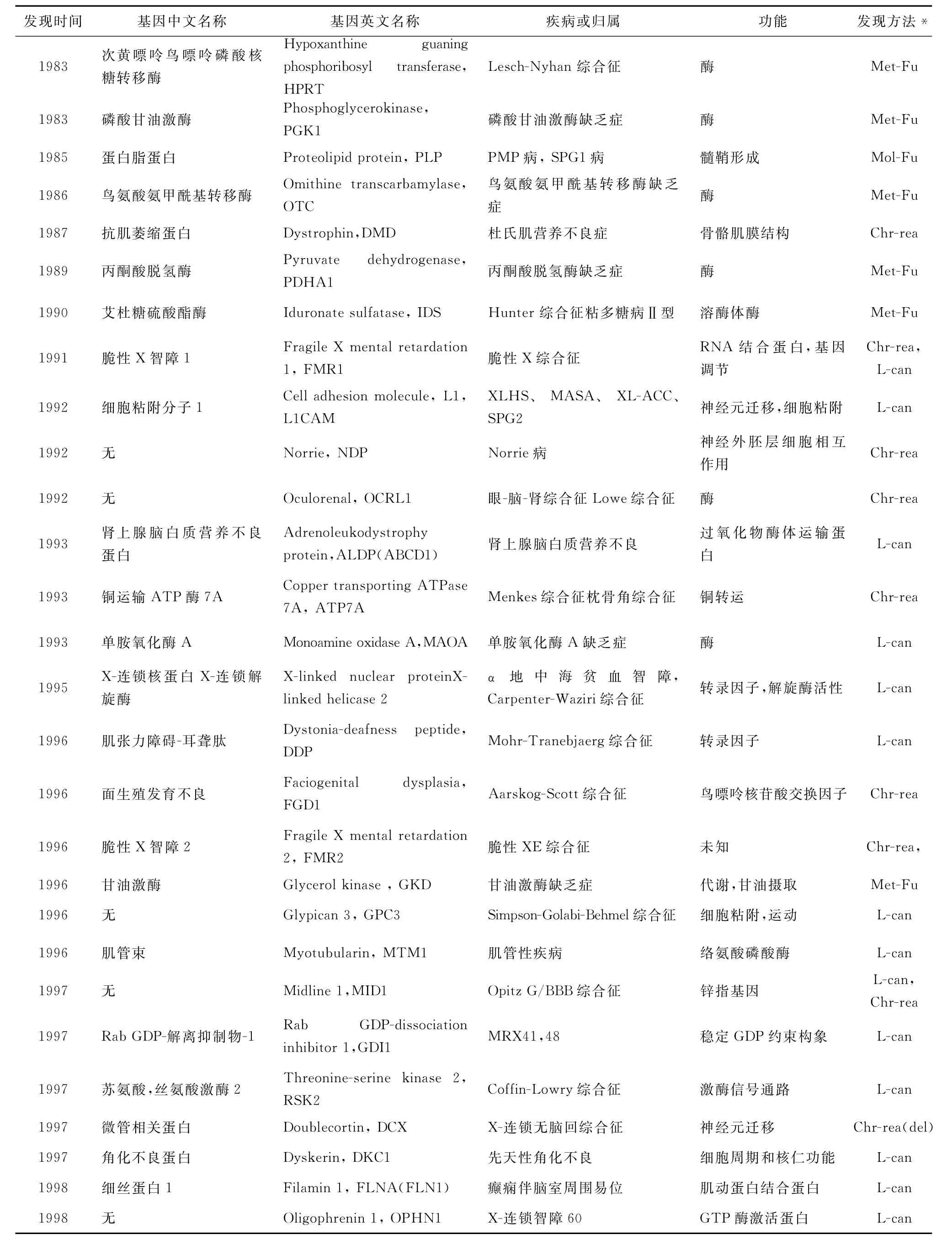
表1 與XLMR相關的基因
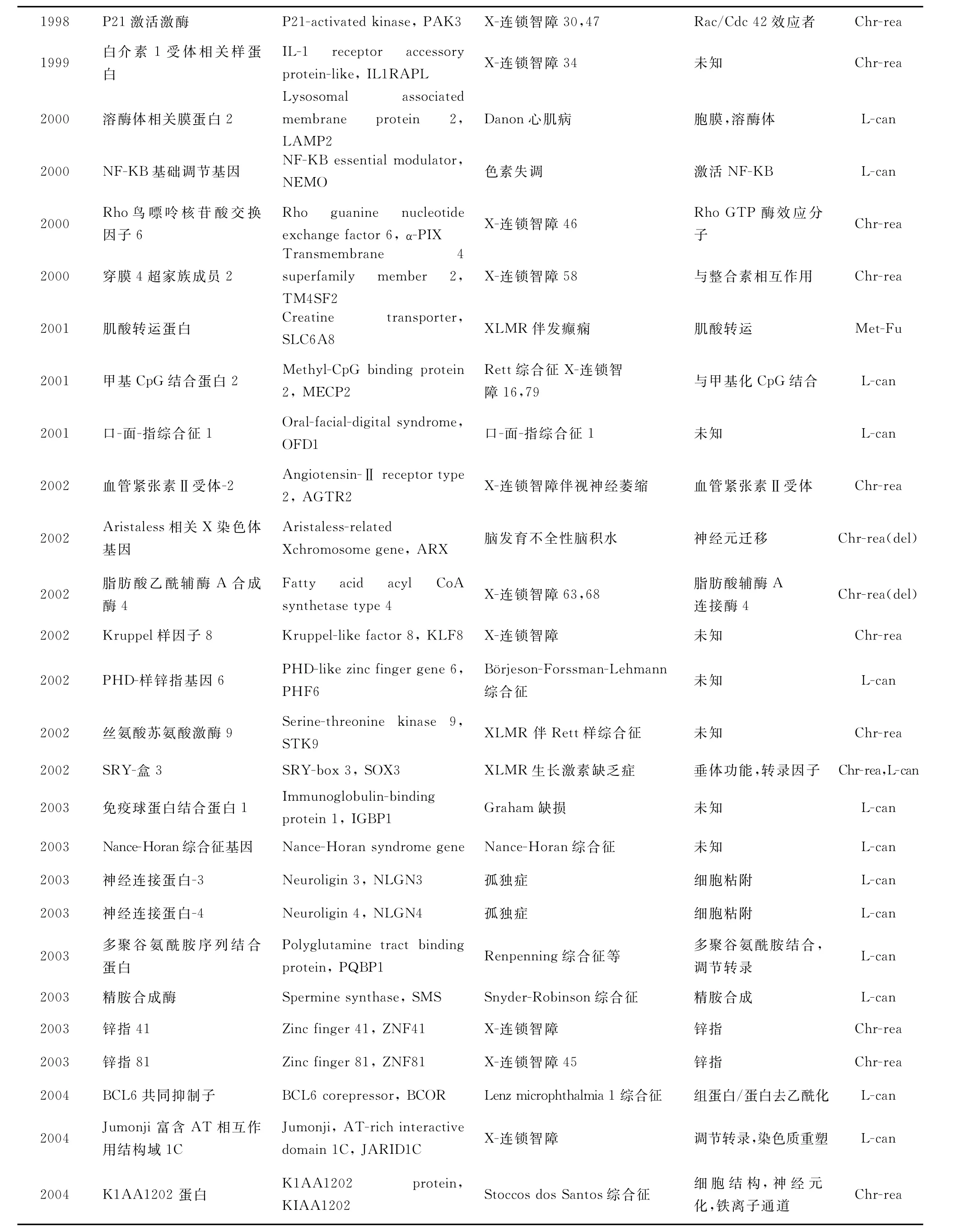
(續表)
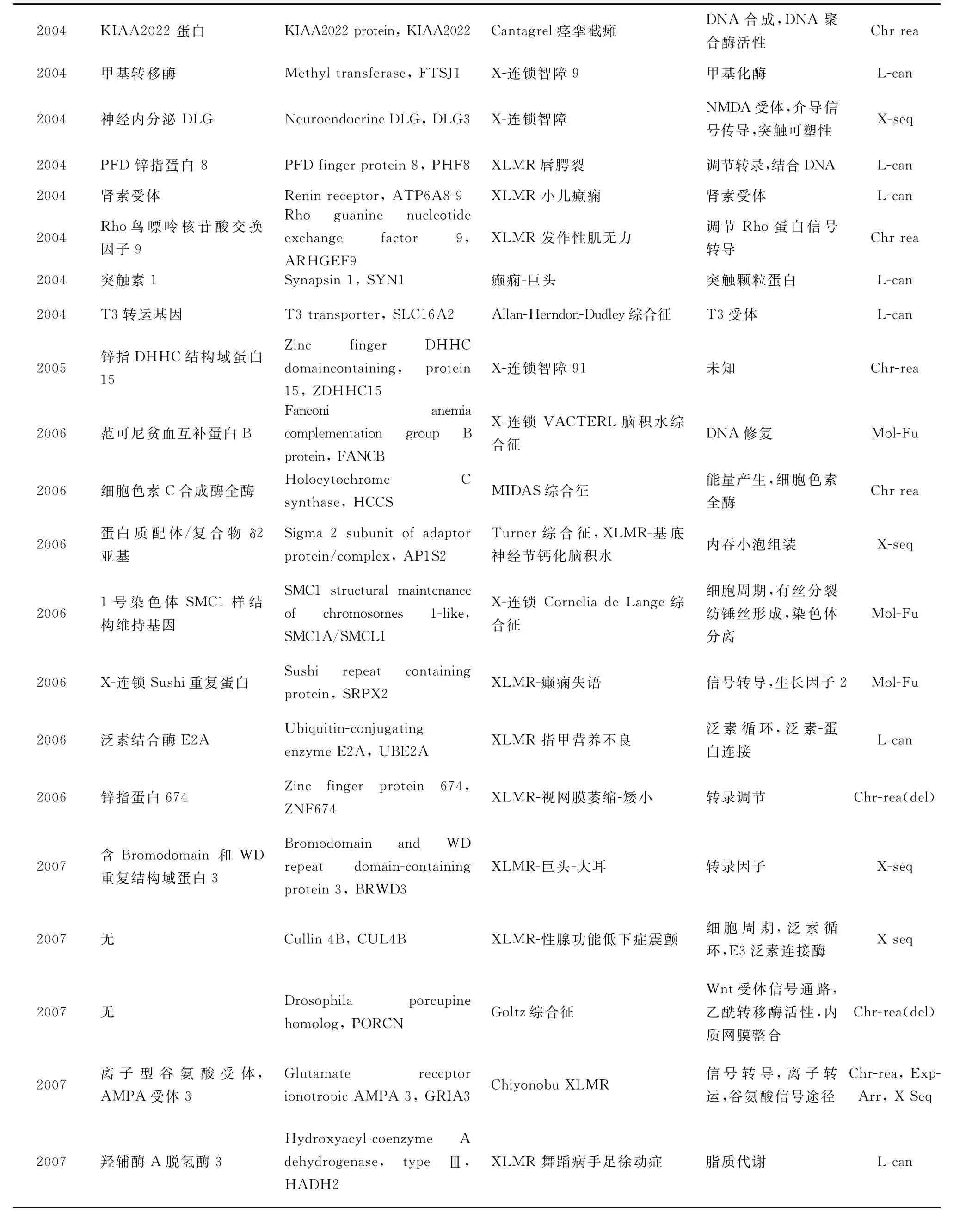
(續表)
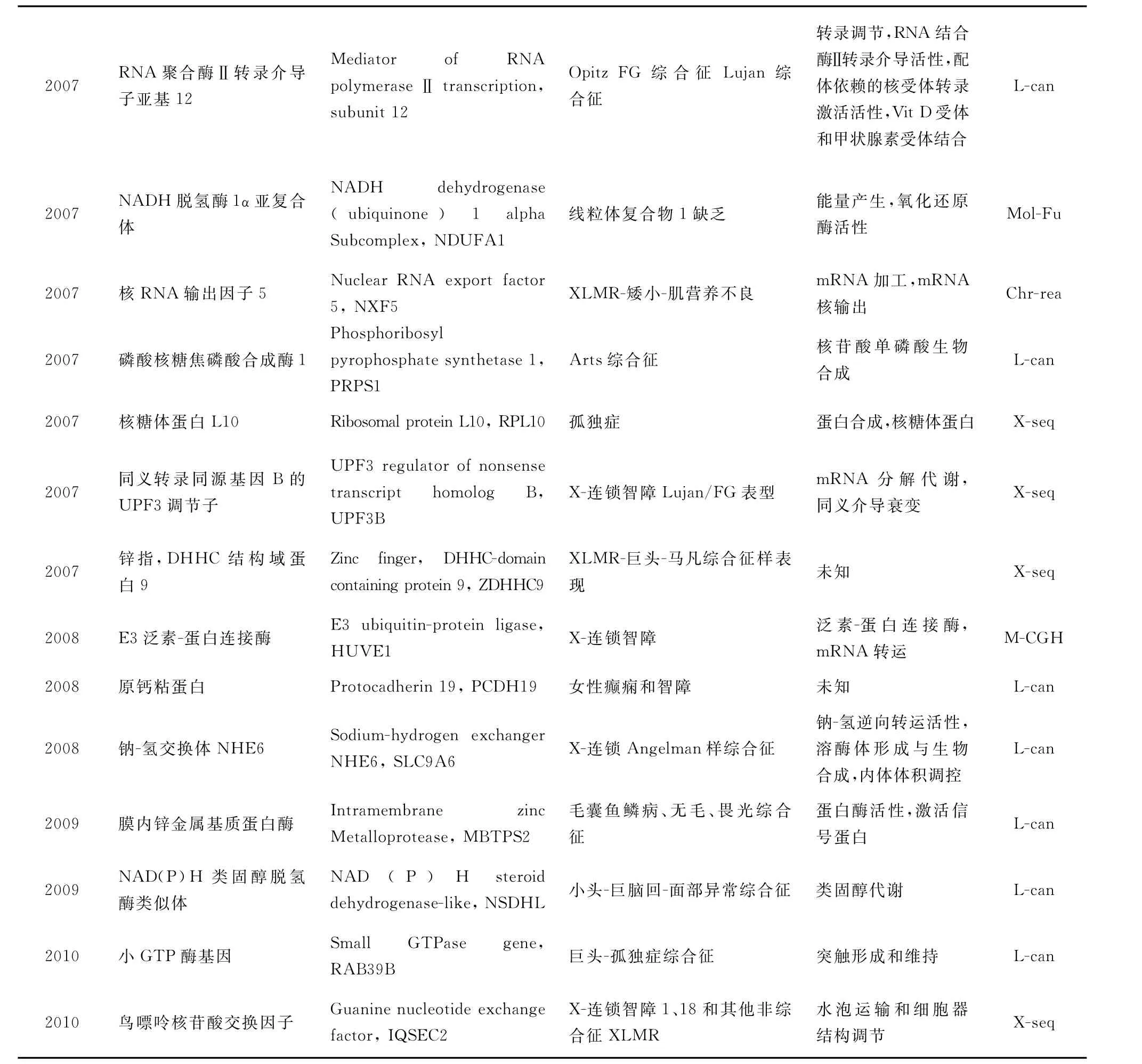
(續表)
3.3 鋅指41基因(ZNF41) 在一個患有X連鎖智障的患者中,Shoichet等[10]發現ZNF41基因出現738C-T突變,從而導致了111位脯氨酸變為亮氨酸(P111L),這些異常同樣也在患者的兄弟中發現。先證者5歲時僅有相當于3歲的智力,表現為語言遲緩、拒絕社會接觸和活動亢進。患者母親的兄弟也患此病,其母親為突變攜帶者。
3.4 神經內分泌DLG3基因(DLG3) 通過對4個中重度X連鎖智障家族中的研究,Tarpey等[11]確認了一個定位于Xq13.1基因上的截短突變。這些家庭的男性患者都表現為中度到重度X連鎖智障,而女性攜帶者則智力正常。DLG3(discs large homolog3)編碼突觸相關蛋白102(SAP102),它是膜相關鳥苷酸激酶蛋白家族成員。神經元SAP102在早期大腦發育過程中表達,形成興奮性突觸的突觸后密集區。
3.5 神經連接蛋白-4基因(NLGN4) 神經連接蛋白是一個蛋白質家族,調節神經元之間的相互作用。神經連接蛋白的功能類似于軸突蛋白家族的配體。NLGN4基因位于Xp22.3,是這個家族中的成員。Laumonnier等[12]報道,在一個大的X連鎖智障法國家庭中,出現孤獨癥或廣泛性發育障礙者,都被確認在NLGN4基因的第五外顯子中發生了2個bp的缺失。該家庭的正常男性未發現此種缺失,女性攜帶者為雜合突變。研究者認為,NLGN4基因突變涉及比較廣泛的表型變化。
3.6 突觸素1(SYN1) Garcia等[13]報道了一個新的X連鎖隱性綜合征,涉及一個4代家系。該家系一些智力正常的男性患有癲癇癥,其他人則表現為癲癇癥、學習困難、巨頭畸型和攻擊性行為。癲癇只在兒童時期發生,個別發生于27歲,并只在有特殊刺激的情況下發生。基因連鎖分析發現致病基因位于Xp11.3-q12,在 MAOB基因和標記物DXS1275之間。通過對10例男性患者和女性攜帶者的SYN1基因測序,研究者確認了一個356色氨酸到終止密碼的突變。
3.7 T3轉運基因(SLC16A2) SLC16A2(solute carrier family 16,member 2)基因定位于Xq13.2,也被稱為MCT8,是一個甲狀腺激素的轉運基因。在對5個不相關的患有重度智障和高三碘甲狀腺素(T3)男孩的研究中,首次發現了該基因的突變。這些患者T3水平異常,表現為發育普遍延遲、肌張力低下、痙攣性截癱、排斥運動、旋轉后眼球震顫、聽力受損和凝視。具有這些表現的患者,應該接受SLC16A2基因測序檢查[14]。
4 XLMR基因的特點
XLMR基因編碼的蛋白可見于細胞所有部位:30%位于核內、28%在細胞質、16%在細胞器、22%在細胞膜、還有一部分不清楚具體的定位。它們的功能可以分為:19%與信號轉導有關、22%與轉錄調節有關、通常是信號級聯反應的最后一步;15%是細胞膜組成成分,同樣也可參與信號轉導。還有一部分XLMR蛋白參與了不同的生物途徑,比如代謝(15%)、DNA 和 RNA 形 成 (6%)、蛋 白 質 合 成(3%)、細胞周期調節和泛素化途徑(7%)。
5 展望
迄今雖已發現近百種XLMR基因,但估計仍然有新的XLMR基因有待發現。潛在XLMR候選基因應該滿足3個優選條件:①在大腦中高表達;②是XLMR已知基因的旁系同源基因,因為這些基因相關的蛋白質也許有相似的功能,比如之前提到的NLGN4和NLGN3;③與已知XLMR蛋白相互作用的蛋白。
智障已經成為影響很多家庭的重要疾病,而遺傳咨詢和對智障的產前診斷又提出了一些敏感的倫理學問題,因此對于智障的分子生物學研究就顯得尤為重要。XLMR作為重要的智障類型更是亟待解決的問題之一,需要更多的研究者為此付出努力。
[1]Lina Basel-Vanagaite.Clinical Approaches to Genetic Mental Retardation[J].IMAJ,2008,10:821-826.
[2]Pietro Chiurazzi,Charles E Schwartz,Jozef Gecz,et al.XLMR genes:update 2007[J].European Journal of Human Genetics,2008,16:422-434.
[3]Malgorzata Zofi a Lisik,Aleksander L,Sieron.X-linked mental retardation[J].Med Sci Monit,2008,14:221-229.
[4]Frints SG,Froyen G,Marynen P,et al.X-linked mental retardation:vanishing boundaries between non-specific(MRX)and syndromic (MRXS)forms[J].Clin Genet,2002,62:423-432.
[5]Ross MT,Grafham DV,Coffey AJ,et al.The DNA sequence of the human X chromosome[J].Nature,2005,434:325-337.
[6]Mnatzakanian GN,Lohi H,Munteanu I,et al.A previously unidentified MECP2 open reading frame defines a new protein isoform relevant to Rett syndrome[J].Nat Genet,2004,36:339-341.
[7]Amir RE,Van den Veyver IB,Wan M,et al.Rett syndrome is caused by mutations in X-linked MECP2,encoding methyl-CpG-binding protein 2[J].Nat Genet,1999,23:185-188.
[8]Hagberg B,Hanefeld F,Percy A,et al.An update on clinically applicable diagnostic criteria in Rett syndrome[J].Euro J Paediatr Neurol,2002,6:293-297.
[9]Salomons GS,van Dooren SJ,Verhoeven NM,et al.X-linked creatine transporter defect:an overview[J].J Inherit Metab Dis,2003,26:309-318.
[10]Shoichet SA,Hoffmann K,Menzel C,et al.Mutations in the ZNF41 gene are associated with cognitive deficits:identification of a new candidate for X-linked mental retardation[J].Am J Hum Genet,2003,73:1341-1354.
[11]Tarpey P,Parnau J,Blow M,et al.Mutations in the DLG3 gene cause non-syndromic X-linked mental retardation[J].Am J Hum Genet,2004,75:318-324.
[12]Laumonnier F,Bonnet-Brilhault F,Gomot M,et al.X-linked mental retardation and autism are associated with a mutation in the NLGN4 gene,a member of the neuroligin family[J].Am J Hum Genet,2004,74:552-557.
[13]Garcia CC,Blair HJ,Seager M,et al.Identification of a mutation in synapsin I,a synaptic vesicle protein,in a family with epilepsy[J].J Med Genet,2004,41:183-187.
[14]Friesema ECH, Grueters A, Biebermann H,et al.Association between mutations in a thyroid hormone transporter and severe X-linked psychomotor retardation[J].Lancet,2004,364:1435-1437.
