HPLC-MS/MS法快速測定淀粉生物粘附材料特異性結合糖類物質的含量
2010-09-01 07:52:14王雪毓李曉璽徐昕榮
質譜學報 2010年5期
向 弘,朱 斌,王雪毓,劉 早,李曉璽,徐昕榮,陳 玲
(1.華南理工大學分析測試中心,廣東廣州 510640; 2.華南理工大學輕工與食品學院,廣東廣州 510640)
HPLC-MS/MS法快速測定淀粉生物粘附材料特異性結合糖類物質的含量
向 弘1,朱 斌1,王雪毓2,劉 早2,李曉璽2,徐昕榮1,陳 玲2
(1.華南理工大學分析測試中心,廣東廣州 510640; 2.華南理工大學輕工與食品學院,廣東廣州 510640)
研究了高效液相色譜-串聯質譜(HPLC-MS/MS)法測定淀粉生物粘附材料與糖殘基特異性結合實驗中,巖藻糖、半乳糖、α-甲基甘露糖苷和 N-乙酰葡萄糖胺4種糖類物質含量的分析方法。將反應后的樣品直接離心過濾后即可利用HPLC-MS/MS進行定性定量分析。以V(乙腈)∶V(水)=80∶20的溶液作為液相色譜流動相,質譜采用電噴霧負離子電離方式掃描,以多反應監測(MRM)模式進行定量分析。在添加水平為1.0~40.0 mg·L-1范圍內,巖藻糖的回收率為111.0%,相對標準偏差(RSD)為6.55%;在添加水平為1.0~80.0 mg·L-1范圍內,半乳糖、α-甲基甘露糖苷和N-乙酰葡萄糖胺的回收率分別為106.5%、99.0%和91.0%,相對標準偏差分別為7.78%、6.64%和5.68%。4種糖類物質的方法檢出限(S/N≥3)分別為0.1 mg·L-1(巖藻糖),1.0 mg·L-1(半乳糖),0.1 mg·L-1(α-甲基甘露糖苷)和0.5 mg·L-1(N-乙酰葡萄糖胺)。該方法可用于評價淀粉粘附材料和糖類物質的特異性結合,為其他領域中糖類物質含量的測定提供可借鑒的方法。
淀粉生物粘附材料;高效液相色譜-串聯質譜(HPLC-MS/MS);糖類物質
生物粘附(bioadhesion)是指天然或合成的高分子物質所具有的能夠粘附到腔道粘液(mucus)或上皮細胞表面的能力[1]。在淀粉生物粘附材料中引入的糖蛋白側鏈能特異性識別細胞表面的糖殘基并與之結合,如 N-乙酰半乳糖胺、N-乙酰葡萄糖胺、半乳糖、果糖及唾液酸等,在細胞識別和粘附反應中起著重要作用[2-3]。因此,通過測定淀粉粘附材料上的糖蛋白與糖類物質特異性結合后剩余的糖量,計算得到能與粘附材料特異性結合的糖,從而可確定淀粉粘附材料與人體胃腸道上皮細胞特異粘附部位。
目前,測定糖類物質含量的最常用方法有比色法、氣相色譜法和高效液相色譜法等。比色法[4]易受到其他可與顯色試劑反應的物質的干擾;氣相色譜法須將樣品衍生化才能進行分析[5];液相色譜法不需要衍生化,但是測定糖常用的示差折光檢測器靈敏度較低,需通過柱前衍生,然后用紫外和熒光檢測提高靈敏度,操作繁雜且常出現異常干擾,影響分析的準確性[5]。高效液相色譜-串聯質譜法(high performance liquid chromatography-tandem mass spectrometry,HPLC-MS/MS)分析物質具有靈敏度高,樣品一般不需衍生化處理的特點。目前國內采用LC/MS法測定糖類物質的報道中,主要是利用單級質譜或多級質譜法對糖類物質做定性研究。徐瑾[6]使用柱前衍生法分別對蘆薈多糖樣品和其水解產物進行衍生,結合LC/MS法檢測,以說明其“結構單元”單糖的組成。孫志偉等[7]利用在線串聯質譜的電噴霧電離模式監測,獲得了油菜花粉多糖中各組分的質譜定性規律。Cappiello等[8]利用親水作用色譜和電噴霧質譜直接研究海洋粘膠中低聚糖的組成情況。Wan等[9]通過LC/MS法研究大氣氣溶膠中三糖以下糖類物質的含量,以[M+NH4]+加合離子作為目標物質的定性定量離子。胡強等[10]建立了LC-MS/MS法測定人尿中乳果糖、甘露醇和乳糖含量,可用于腸通透性評價。而利用 HPLCMS/MS法直接進行糖類物質定量分析的報道較少。
用LC/MS法進行定量分析時,不采用總離子色譜圖,而是采用與待測組分相對應的特征離子得到的質量色譜圖或多離子監測色譜圖。此時,不相關的組分將不出峰,可以減少組份間的互相干擾。LC/MS定量的最好方法是采用串聯質譜(MS/MS)的多反應監測(MRM)模式。MRM技術是基于已知信息或假定信息,設定質譜檢測規則,對符合規則的離子進行信號記錄,去除大量不符合規則的離子信號干擾,從而得到質譜信息的一種數據獲取方式。具體地說,即是根據母離子質量數和特征碎片離子質量數,選擇母離子/特征碎片離子對,允許符合設定的母離子進入碰撞室,碰撞完成后,只記錄設定特征碎片離子信號。通過母離子和特征碎片離子的兩次選擇,去除干擾離子,降低化學背景,提高靈敏度[11]。
本工作選擇4種細胞表面含有的糖類物質,巖藻糖、半乳糖、α-甲基甘露糖苷和 N-乙酰葡萄糖胺,利用HPLC-MS/MS的MRM模式作為定量研究手段,直接測定未與淀粉粘附材料特異結合的剩余糖量,可用于評價淀粉生物粘附材料和細胞表面糖類物質的特異性結合。
1 材料與方法
1.1 試劑與儀器
乙腈(色譜純):德國Merk公司產品;水:超純水;巖藻糖、半乳糖、α-甲基甘露糖苷和 N-乙酰葡萄糖胺標準品:美國 Vector Laboratories公司產品。
Bruker Esquire HCT PLUS大容量離子阱質譜儀:德國Bruker公司產品,配有電噴霧離子源(ESI);1100系列高效液相色譜儀:美國Agilent公司產品,配有四元泵、在線真空脫氣機、柱溫箱和自動進樣器。
1.2 標準溶液配制
準確量取一定體積的巖藻糖、半乳糖、α-甲基甘露糖苷和 N-乙酰葡萄糖胺標準品,用超純水分別配制成濃度為1 g·L-1的標準儲備液,搖勻,置于2~8℃,保存備用。
1.3 樣品處理
將一定質量的淀粉生物粘附材料分別與已配制成一定濃度的巖藻糖、半乳糖、α-甲基甘露糖苷和N-乙酰葡萄糖胺溶液反應2 h,反應后將反應液以8 000 r·min-1高速離心3 min,取上清液,用0.2μm濾膜過濾后,進行液相色譜-質譜分離和檢測。
1.4 試驗條件
1.4.1 色譜條件 色譜柱:Agilent ZORBAX N H2(250 mm×4.6 mm×5μm);流動相:V(乙腈)∶V(水)=80∶20的溶液;流速:0.6 mL· min-1;柱溫:常溫;進樣量:10μL。
1.4.2 質譜條件 電噴霧離子源負電子掃描(ESI-),多反應監測模式(MRM);毛細管電壓: 4 000 V;霧化氣壓力:0.138 MPa;干燥氣流量: 10 L·min-1;干燥氣溫度:250℃。
2 結果與討論
2.1 質譜分析
取20 mg·L-1的巖藻糖、半乳糖、α-甲基甘露糖苷和 N-乙酰葡萄糖胺標準溶液,分別將其標準溶液采用直接注射進樣法注入離子源中,在電噴霧負離子電離方式下進行母離子全掃描,對質譜條件進行優化,質譜條件主要是掃描范圍、毛細管出口電壓和錐孔電壓,得到目標物質的[M-H]-準分子離子峰,確定巖藻糖、半乳糖、α-甲基甘露糖苷和 N-乙酰葡萄糖胺的準分子離子峰分別是 m/z163、179、193和220。分別以上述離子為母離子進行二級質譜分析,采用全掃描的二級離子質譜圖得到碎片離子信息,然后再對得到的目標物質的二級質譜碎裂電壓進行優化。為了增加檢測的靈敏度,以MRM模式調諧二級質譜碎裂電壓,即MRM碰撞能量,使每種糖類物質的母離子與特征碎片離子產生的離子對相對強度達到最大時為最佳。4種糖類物質的一級及二級質譜圖示于圖1,優化后的巖藻糖、半乳糖、α-甲基甘露糖苷和 N-乙酰葡萄糖胺的質譜分析條件列于表1。
由圖1(a2)可見,巖藻糖二級質譜的主要特征碎片離子是 m/z145和 m/z89,各特征碎片離子歸屬如下:m/z145為巖藻糖分子離子的失水峰[M-H2O-H]-;m/z89為巖藻糖分子離子裂解后所產生的丁二醇分子的碎片離子峰。
由圖1(b2)可見,半乳糖二級質譜的主要特征碎片離子是 m/z161、143和89,各特征碎片離子歸屬如下:m/z161為半乳糖分子離子的失水峰[M-H2O-H]-;m/z143為半乳糖分子離子失去2個水的離子峰[M-2H2O-H]-; m/z89為半乳糖分子裂解后所產生的丁二醇分子的碎片離子峰。
由圖1(c2)可見,α-甲基甘露糖苷二級質譜的主要特征碎片離子是m/z161、145和101,各特征碎片離子歸屬如下:m/z161為α-甲基甘露糖苷分子經碰撞后丟失甲基后的失水峰[MCH3-H2O]-;m/z145為α-甲基甘露糖苷分子經碰撞后丟失甲氧基后的失水峰[M-OCH3-H2O]-;m/z101為α-甲基甘露糖苷分子裂解后所產生的戊烯二醇分子的碎片離子峰。
由圖1(d2)可見,N-乙酰葡萄糖胺二級質譜的主要特征碎片離子是m/z119和101,各特征碎片離子歸屬如下:m/z119為α-甲基甘露糖苷分子裂解后所產生的戊三醇分子的碎片離子峰; m/z101為戊三醇分子離子進一步失水后所產生的戊烯二醇分子的碎片離子峰。
由以上4種糖類物質二級質譜圖碎片離子分析可知,糖類物質分子在二級電離過程中易丟失水分子形成失水峰,并且也較易獲得穩定度較高的多元醇分子或烯醇分子的碎片離子。巖藻糖、半乳糖、α-甲基甘露糖苷和 N-乙酰葡萄糖胺二級質譜圖中,離子強度最高的碎片離子分別為m/z89、89、101和119,因此以 m/z89(163), m/z89(179),m/z101(193)和m/z119(220)分別作為對應糖類物質定量分析時的定量離子。

表1 巖藻糖、半乳糖、α-甲基甘露糖苷和N-乙酰葡萄糖胺的質譜分析條件Table 1 The analysis parameters of mass spectrum of fucose,galactose,α-methyl mannoside andN-acetylglucosamine
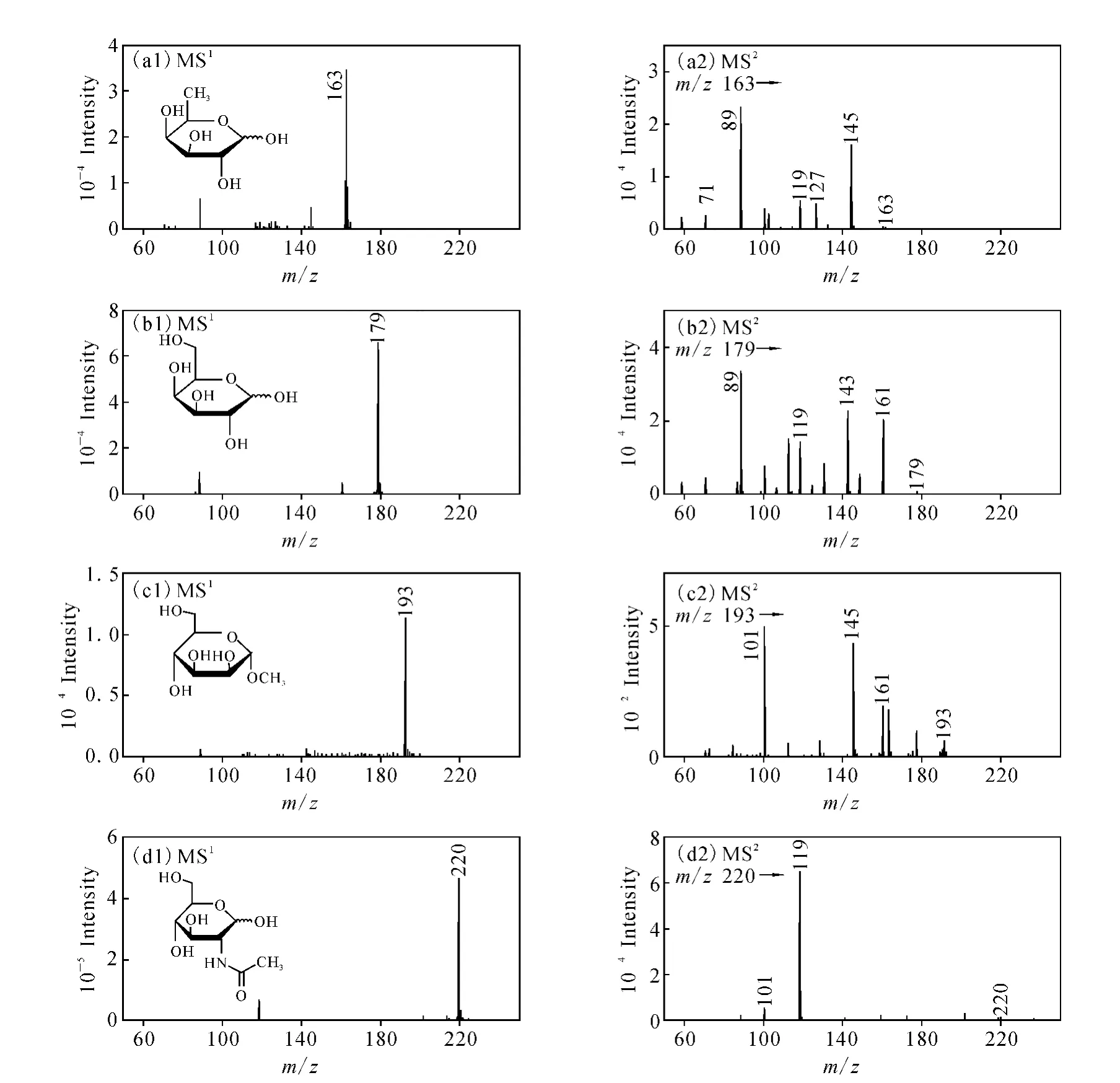
圖1 4種糖類物質的一級和二級質譜圖(a1)、(a2)巖藻糖;(b1)、(b2)半乳糖;(c1)、(c2)α-甲基甘露糖苷;(d1)、(d2)N-乙酰葡萄糖胺Fig.1 ESI(-)-MS and ESI(-)-MS/MS product-ion mass spectra of glucide (a1),(a2)fucose;(b1),(b2)galactose;(c1),(c2)α-methyl mannoside;(d1),(d2)N-acetylglucosamine
2.2 液相分析
分離糖類物質所采用的色譜柱主要有兩類,即氨基柱和糖柱[5]。糖柱采用的是水作為流動相,在質譜分析階段純水會影響質譜的離子強度,而氨基柱使用了乙腈-水作為流動相,因此最終采用氨基色譜柱。
流動相中乙腈含量的多少直接影響分離效果。增加有機相有利于4種糖類物質組分間的分離;相反,增加水相比例有利于糖類的溶解,但不利于各組分間的分離。經多次實驗,最終確定流動相為V(乙腈)∶V(水)=80∶20的溶液。巖藻糖、半乳糖、α-甲基甘露糖苷和 N-乙酰葡萄糖胺標準物質的提取子離子色譜圖示于圖2,以m/z89(163),m/z89(179),m/z101(193)和m/z119(220)分別作為4種糖類物質的提取離子。從圖2可以看出,4種糖類物質得到較好的分離,各組分分析時間短,均可在8 min內完成。因此本方法可用于單獨快速測定4類糖類物質的特異性結合實驗,同時也可滿足混合樣品的分析,樣品中各糖類物質的提取子離子色譜圖示于圖3。
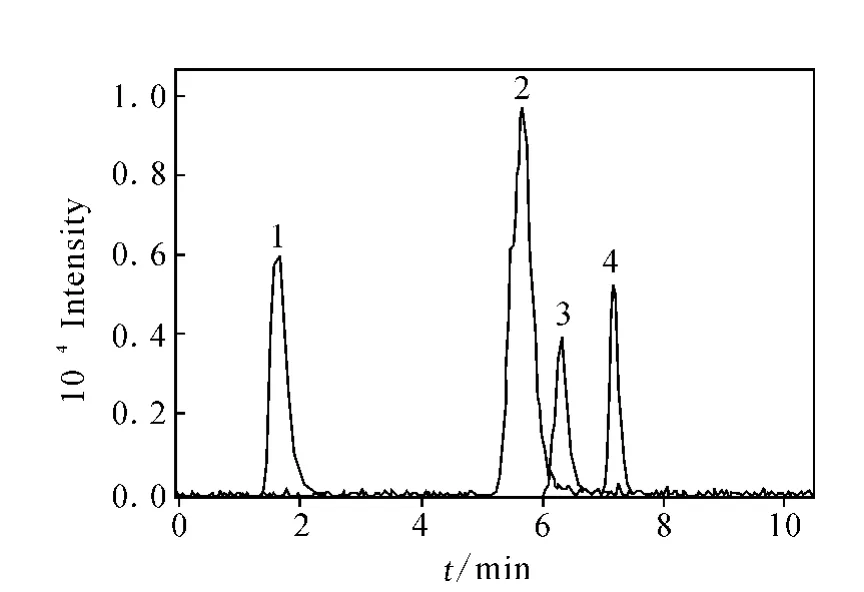
圖2 4種糖類標準物質的提取子離子色譜圖Fig.2 The extracted ion chromatogram of standard glucide
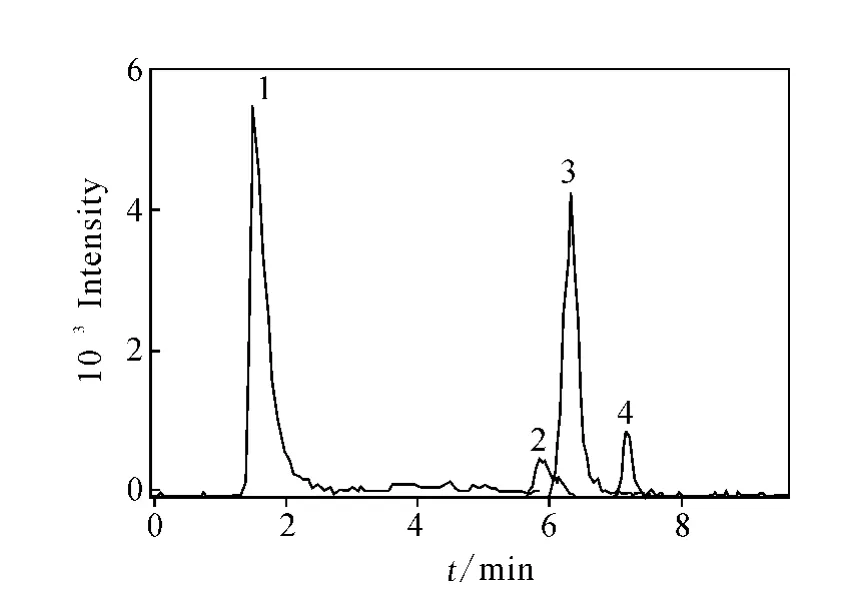
圖3 樣品中4種糖類物質的提取子離子色譜圖Fig.3 The extracted ion chromatogram of samples
2.3 線性范圍
取巖藻糖、半乳糖、α-甲基甘露糖苷和 N-乙酰葡萄糖胺的標準儲備液,分別用超純水配制成標準工作溶液,巖藻糖標準工作溶液的濃度為1.0、10.0、20.0、30.0和40.0 mg·L-1,半乳糖、α-甲基甘露糖苷和 N-乙酰葡萄糖胺標準工作溶液的濃度為1.0、10.0、20.0、40.0、60.0和80.0 mg·L-1。
以標準濃度為 x軸,峰面積為 y軸,進行回歸分析,經“1/X”權重可得回歸方程:巖藻糖回歸方程為 y=-3.27×103+4.35×103x,r= 0.994 5;半乳糖回歸方程為 y=2.96×103+ 1.20×103x,r=0.999 6;α-甲基甘露糖苷回歸方程為 y= -1.03×102+1.18 ×103x, r=0.998 7;N-乙酰葡萄糖胺回歸方程為 y= -1.03×104+4.63×103x,r=0.999 3。實驗結果表明,在1.0~40.0 mg·L-1范圍內,巖藻糖具有良好的線性關系,半乳糖、α-甲基甘露糖苷和 N-乙酰葡萄糖胺在1.0~80.0 mg·L-1范圍內同樣也具有良好的線性關系,它們都符合定量的要求。
2.4 回收率和精密度試驗
用標準加入法進行回收率實驗,對方法的準確度和精密度進行考察。在空白樣品中精密加入巖藻糖、半乳糖、α-甲基甘露糖苷和 N-乙酰葡萄糖胺的標樣溶液進行加標回收試驗,結果列于表2。
2.5 靈敏度
依據樣品的處理方法和測試條件,采用質譜進行定量分析時,4種糖類物質的方法檢出限(S/N≥3)分別為0.1 mg·L-1(巖藻糖)、1.0 mg·L-1(半乳糖)、0.1 mg·L-1(α-甲基甘露糖苷)和0.5 mg·L-1(N-乙酰葡萄糖胺)。
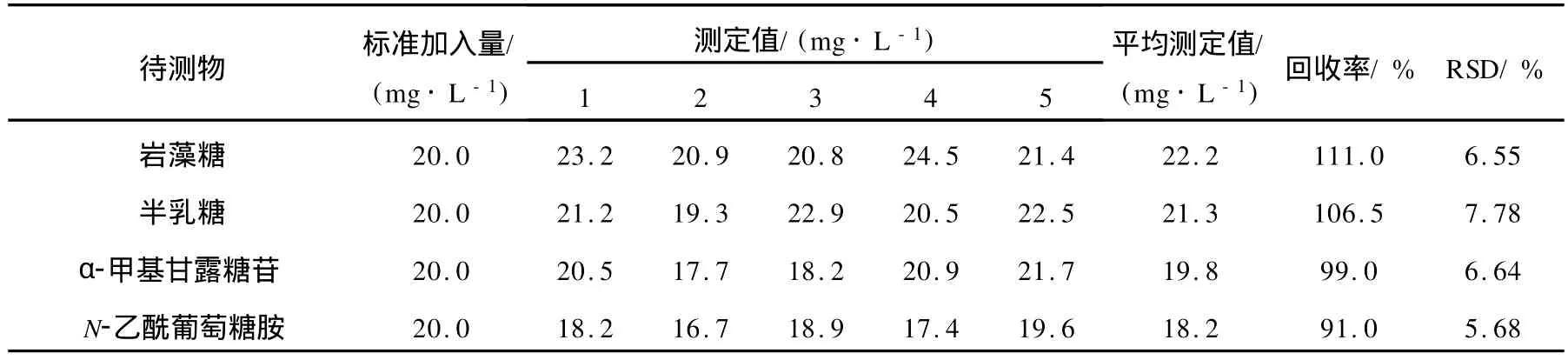
表2 回收率與精密度測定結果(n=5)Table 2 Results of tests for recovery and precision of the method(n=5)
3 結 論
本工作建立了快速靈敏的檢測與淀粉生物粘附材料特異性結合的4種細胞表面含有的糖類物質含量的高效液相色譜-串聯質譜的分析方法,通過離子阱質譜的多級質譜功能對樣品進行定性及精確定量分析。本方法樣品直接離心過濾后即可進樣,大大簡化了樣品處理步驟,提高分析效率,而且靈敏度高,分析時間短,回收率為111.0%~91.0%,重現性好,能滿足生物樣品分析方法的有關要求,為淀粉生物粘附材料與細胞表面糖類物質的特異性結合評價提供了可靠的分析方法,同時也為其他領域中糖類物質含量的測定提供了可借鑒的方法。
[1]王 健,畢殿洲.生物粘附材料的研究進展[J].沈陽藥科大學學報,2002,19(5):373-380.
[2]HAL TNER E,EASSON J H,L EHR C M.Lectins and bacterial invasion factors for controlling endo-and transcytosis of bioadhesive drug carrier systems[J].Eur J Pharm Biopharm,1997,44:3-13.
[3]CLARK M A,J EPSON M A,SIMMONS N L,et al.Differential expression of lectin-binding sites defines mouse intestinal Mcells[J].J Histochem Cytochem,1993,41:1 679-1 687.
[4]林炎坤.常用的幾種蒽酮比色定糖法的比較和改進[J].植物生理學通報,1989,(4):53-55.
[5]王 靜,王 晴,向文勝.色譜法在糖類化合物分析中的應用 [J].分析化學,2001,29(2): 222-227.
[6]徐 瑾.高效凝膠滲透色譜及高效液相色譜和電噴霧-離子阱質譜法聯用測定構成蘆薈多糖的單糖[J].理化檢測:化學分冊,2008,44(12):1 133-1 136.
[7]孫志偉,劉凌君,戶寶軍,等.1-(2-萘基)-3-甲基-5-吡唑啉酮衍生試劑的制備及其在高效液相色譜-質譜法測定糖類化合物中的應用[J].色譜, 2008,26(2):200-205.
[8]CAPPIELLOA A,TRUFELLIA H,FAMIGLINIA G,et al.Study on the oligosaccharides composition of the water soluble fraction of marine mucilage by electrospray tandem mass spectrometry [J].Water Research,2007,41:2 911-2 920.
[9]WAN E C H,YU J Z.Determination of sugar compounds in atmospheric aerosols by liquid chromatography combined with positive electrospray ionization mass spectrometry[J].J Chromatogr A, 2006,1 107:175-181.
[10]胡 強,徐紅兵,李水軍,等.液相色譜-串聯質譜法測定尿中乳果糖、甘露醇和乳糖含量[J].中國現代醫學雜志,2008,18(13):1 810-1 817.
[11]BRUNO D,RUEDI A.mass spectrometry and protein analysis[J]. Science,2006,312: 212-217.
Rapid Detection of Glucide Specifically Linking with Starch Bioadhesive Materials by HPLC-MS/MS
XIANG Hong1,ZHU Bin1,WAN G Xue-yu2,LIU Zao2,LI Xiao-xi2,XU Xin-rong1,CHEN Ling2
(1.A nalytical and Testing Center,South China University ofTechnology,Guangzhou510641,China; 2.School of Light Chemistry and Food Science,South China University ofTechnology,Guangzhou510641,China)
A method was developed in determining 4 kinds of glucides which were fucose,galactose,α-methyl mannoside andN-acetylglucosamine,in experiment of adhesive property between starch bioadhesive materials and sugar residues by high performance liquid chromatography-tandem mass spectrometry(HPLC-MS/MS).The sample was filtrated and analyzed by HPLC-MS/MS.Analyte was separated by liquid chromatography with a mobile phase consisting ofV(acetonitrile)∶V(water)=80∶20.The mass spectrometer was opera-ted at the negative ion mode via electrospray ionization(ESI)source.The quantitative analysis was determined by the multiple reactions monitoring(MRM).Linear relationship between values of peak area of detection signals and concentration of fucose are obtained in the ranges of 1.0—40.0 mg·L-1,while relationships of galactose,α-methyl mannoside andN-acetylglucosamine are obtained in the range of the linear ranges 1.0—80.0 mg·L-1.The recovery rate of fucose is 111%,and the RSD is 6.55%.The recoveries rates of galactose, α-methyl mannoside andN-acetylglucosamine are 106.5%,99.0%and 91.0%,the RSDs are 7.78%,6.64%and 5.68%,respectively.Limits of detection(S/N≥3)for fucose,galactose,α-methyl mannoside andN-acetylglucosamine are 0.1,1.0,0.1 and 0.5 mg·L-1, respectively.The method is proved to be rapid,reliable and sensitive,which is suitable for estimating the specific linking between starch bioadhesive materials and glucides.The proposed method is also suitable for rapid determination of glucides components in other complex system.
starch bioadhesive materials;high performance liquid chromatography-tandem mass spectrometry(HPLC-MS/MS);glucides
O 657.63
A
1004-2997(2010)05-291-06
2009-11-09;
2010-03-29
國家自然科學基金項目(20606014);國家支撐項目(2006BAD27B04)資助
向 弘(1980~),女(土家族),工程師,從事質譜儀測試方面研究。E-mail:hxiang@scut.edu.cn
陳 玲(1964~),女(漢族),博士,教授,從事生物材料方面研究。E-mail:felchen@scut.edu.cn
