原花青素脂質體的制備條件優化
2010-08-08 06:37:14姚薇薇
東北農業大學學報 2010年9期
胡 博,姚薇薇,劉 寧
(乳品科學教育部重點實驗室,東北農業大學食品學院,哈爾濱 150030)
原花青素(Procyanidins,PC,曾用英文名Proanthocyanidins,Pycnogenol)是一大類多酚化合物的總稱,由不同數目的黃烷-3-醇或黃烷-3,4-二醇聚合而成。按聚合度大小,二至四聚體稱為低聚原花青素(Oligomeric proanthocyanidins,OPCs),五聚體以上稱為多聚原花青素(Polymers procyanidins,PPCs)[1-2]。近代研究發現,原花青素具有極強的清除自由基能力和顯著的抗高血壓[3-4]、抗動脈粥樣硬化、抗腫瘤防癌等生理功能,其清除自由基的能力是VC的20倍、VE的50倍[5-6],以安全低毒、高效、高生物利用率而著稱[7],其在食品添加劑、保健品、藥物和化妝品等領域得到了廣泛的應用。由此可見,對原花青素進行研究,有利于更好地利用該寶貴資源。
原花青素的水溶性較好[8],但穩定性較差,易受外界條件影響[9],如易氧化,對光、熱、pH值敏感等[10-11],這使其應用受到限制。因此,必須對它進行保護。目前研究較多的是原花青素微膠囊,并且已經進行了工業化生產,但是其粒徑較大,不利于人體吸收,如何在有效保護原花青素的同時減小其粒徑一直是個難題。采用脂質體包埋可以解決這一難題[12],脂質體粒徑小,具有靶向性,有利于吸收,制備工藝簡單,具有廣泛的應用前景。制備脂質體的方法很多,其中逆相蒸發法制備的大單層脂質體具有較大的水性空間,更適合對水溶性物質的包埋[13]。任文霞等在進行茶多酚脂質體制備方法的篩選時,發現逆相蒸發法制備的茶多酚脂質體包埋率較高,可達48.9%[14]。周威比較了在相同條件下應用逆相蒸發法和其他方法制備了溶菌酶脂質體,經過研究他發現逆相蒸發法的包埋率最高,適用于工業生產[15]。
本試驗以大豆卵磷脂、膽固醇為膜材,采用逆相蒸發法制備原花青素脂質體。以脂質體包埋的形式來增強原花青素的穩定性,通過試驗研究,對原花青素脂質體的制備條件進行了優化。
1.3 方法
1 材料與方法
1.1 試劑
原花青素,純度為95%(購自山東臨沂泰豪國際貿易有限公司);大豆卵磷脂(購自上海源聚生物科技有限公司);膽固醇(購自天津市博迪化工有限公司);無水乙醇、正丁醇、鹽酸及其他試劑均為分析純。
1.2 儀器
ZFQ85B旋轉蒸發儀(購自上海醫械專機廠);KQ32000DB型數控超聲波清洗器(購自上海昆山超聲儀器有限公司);SYNERGY超純水儀(購自美國Millipore公司);AL204精密電子天平(購自瑞士梅特勒-托利多公司);UV-2401PC紫外可見分光光度計(購自日本島津公司);DelsaNano C型粒度分析儀(購自美國貝克曼庫爾特有限公司),DelsaNano C型Zeta電位分析儀(購自美國貝克曼庫爾特有限公司);透射電子顯微鏡(購自日本日立H-7650)。
1.3.1 逆相蒸發法制備空白脂質體
取總量為0.4 g,一定比例的大豆卵磷脂、膽固醇置于梨形燒瓶中,加入體積比為1:1的乙醚和乙醇溶解,振搖,再加入一定濃度的2 mL磷酸緩沖液,震蕩混合均勻,超聲5 min,旋轉至干成膜,加入磷酸緩沖溶液,使膜溶解并充分水合,將所得粗混懸液過0.45 μm濾膜,得到空白脂質體。
1.3.2 逆相蒸發法制備原花青素脂質體
取總量為0.4 g,一定比例的大豆卵磷脂、膽固醇置于梨形燒瓶中,加入體積比為1:1的乙醚和乙醇溶解,振搖,再加入一定濃度的2 mL磷酸緩沖原花青素溶液,震蕩混合均勻,超聲5 min,旋轉至干成膜,加入磷酸緩沖溶液,使膜溶解并充分水合,將所得粗混懸液過0.45 μm濾膜,得到原花青素脂質體混懸液。
1.3.3 原花青素的測定
取不同濃度原花青素對照品的樣液0.5 mL,加入到20 mL的具塞試管中,然后加入5 mL正丁醇-鹽酸(體積比95:5)溶液,搖勻。打開塞子放入97℃恒溫水浴中,3 min后塞緊塞子,加熱40 min后,打開塞子,冷卻5 min。于546 nm處測定吸光值,繪制標準曲線。
1.3.4 原花青素脂質體包埋率的測定
總原花青素含量:取等量原花青素脂質體和空白脂質體于試管中,加入適量無水乙醇破乳,15 000 r·min-1離心30 min,取上清液,定容至25 mL,以離心后的空白脂質體作對照,用正丁醇-鹽酸法測定制備樣品中總原花青素的含量。
游離的原花青素含量:吸取等量原花青素脂質體和空白脂質體于離心管中,15 000 r·min-1離心30 min,取上清液,定容至25 mL,以離心后的空白脂質體作對照,用正丁醇-鹽酸法測定制備樣品中游離原花青素的含量。
包埋率計算公式為:包埋率(%)=(添加原花青素總含量+游離原花青素的量)/添加原花青素總量×100%
1.3.5 脂質體顯微形態觀察
將脂質體混懸液在透射電子顯微鏡下進行觀察。透射電子顯微鏡采用磷鎢酸負染法進行,即將脂質體稀釋一定倍數后滴至專用銅網上,靜止吸附3 min,用濾紙吸干銅篩邊緣多余的液體樣品,然后放在一滴用2.0%磷鎢酸溶液上進行復染,用濾紙吸干銅篩邊緣多余的染色液,自然晾干后放入透射電子顯微鏡下進行觀察。
1.3.6 脂質體粒徑的測定
依次取適量的脂質體混懸液,用DelsaNano C粒度分析儀測定原花青素脂質體的粒徑。每個樣品重復運行3次取平均值。
1.3.7 脂質體Zeta電位的測定
依次取適量的脂質體混懸液放入樣品池中,用DelsaNano C電位儀測定脂質體的電位。每個樣品測定3次取平均值。
2 結果與分析
2.1 標準曲線的繪制
圖1為原花青素不同濃度組成標準曲線。由圖可知,在測定范圍內,原花青素的濃度與吸光值有良好的線性關系,線性方程為y=1.4011x-0.0076,以此標準曲線計算原花青素脂質體的包埋率。
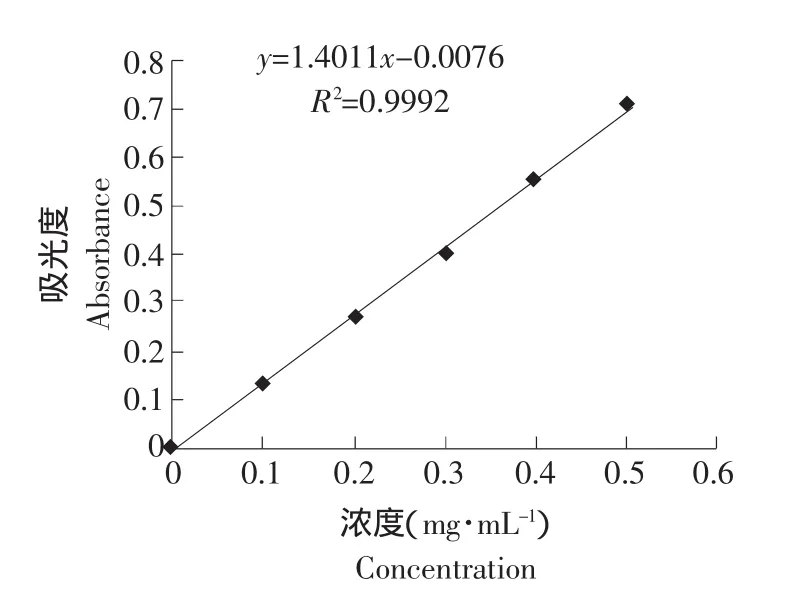
圖1 原花青素濃度標準曲線Fig.1 Standard curve of procyanidine concentration
2.2 膽固醇與卵磷脂的不同比例對包埋率的影響
膽固醇與卵磷脂是脂質體形成不可缺少的膜材,二者比例是影響脂質體包埋率的主要因素。卵磷脂是脂質體的主要膜材,加入膽固醇可以改變其相變溫度,對脂質膜的流動性產生雙向調節功能。根據國內外已有文獻及預實驗結果,膽固醇與卵磷脂物質的量比在1:1~1:5包埋率較高。在一定的比例范圍內,如果膽固醇的比例過大,組成脂質體的卵磷脂添加量太少,脂質體膜的形成就會困難,而且不牢固,而由此形成的脂質體膜親水性太強,膜也容易破壞。固定其他組分的比例不變,僅改變膽固醇和卵磷脂的比例,制備原花青素脂質體,包埋率隨兩者比例的變化情況見圖2。
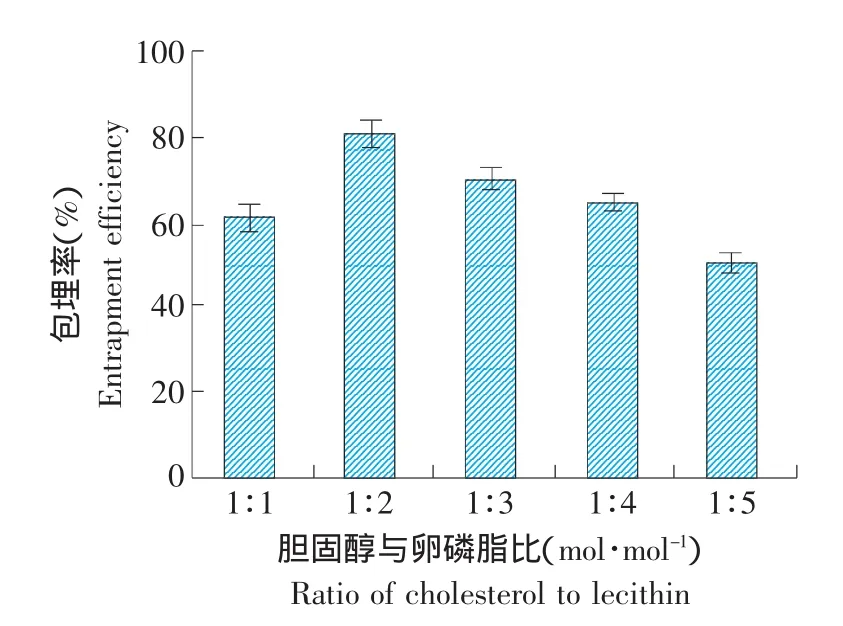
圖2 膽固醇與卵磷脂比對包埋率的影響Fig.2 Effect of the ratio of cholesterol to lecithin on entrapment efficiency of procyanidine liposomes
由圖2可知,隨著卵磷脂的增加,原花青素脂質體的包埋率先增加后減小。當膽固醇與卵磷脂摩爾為1:2時,原花青素脂質體包埋率最高,為80.5%±3.4%。
2.3 原花青素添加量對包埋率的影響
水溶性藥物被包埋于內核水相中,經常在脂質體水相和油相間重新分配,容易引起藥物的泄露,所以原花青素的添加量直接影響脂質體的包埋率。選擇原花青素與卵磷脂的不同比例,根據預實驗當其比例在1:10~1:50時包埋率較高。固定其他組分的比例不變,只改變原花青素與卵磷脂的比例,制備原花青素脂質體,包埋率隨兩者比例的變化情況見圖3。
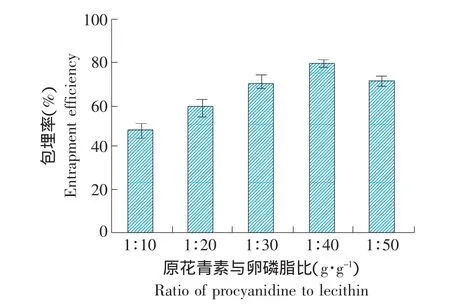
圖3 原花青素與卵磷脂比對包埋率的影響Fig.3 Effect of the ratio of procyanidine to lecithin on entrapment efficiency of procyanidine liposomes
由圖3可知,隨著卵磷脂的增加,脂質體的包埋率升高,但藥脂比大于1:40后,包埋率開始降低。當藥脂比為1:40時,包埋率達到最高值。
2.4 水溶液與有機溶劑不同比例對包埋率的影響
通過逆相蒸發法可以得到大單層脂質體,脂質體有較大的水性空間,所以選擇適當的水溶液與有機溶劑體積比可使內相體積增加,通常選擇在1:2~1:6范圍內。固定其他組分的比例不變,改變水溶液與有機溶劑比例,制備原花青素脂質體,包埋率隨兩者比例的變化情況見圖4。
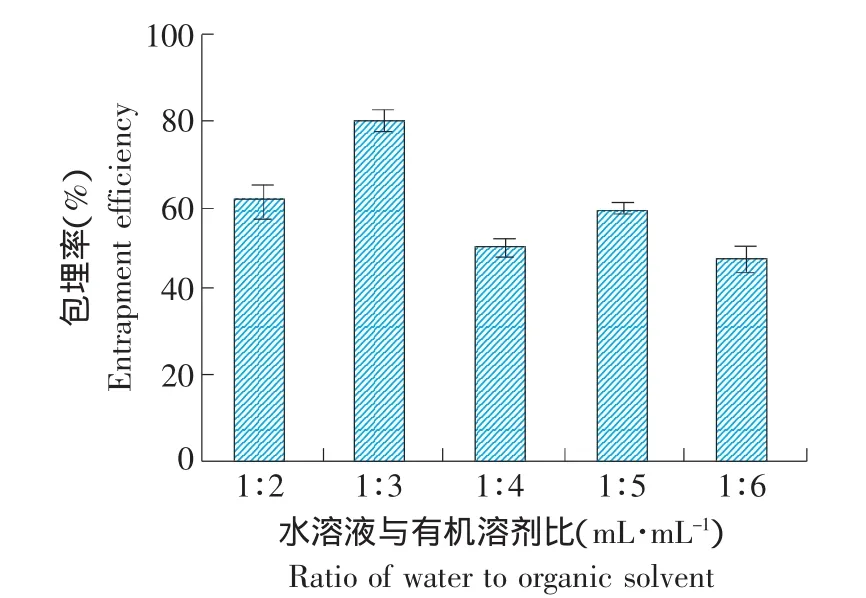
圖4 水溶液與有機溶劑比對包埋率的影響Fig.4 Effect of the ratio of water to organic solvent on entrapment efficiency of procyanidine liposomes
由圖4可知,當比例為1:3時,包埋率最高。在制備過程中,水溶液與有機溶劑的比例影響脂質材料的溶解度,從而影響脂質體的包埋率。
2.5 溫度對包埋率的影響
制備過程中,水浴溫度高時脂質的成膜速度快,卵磷脂和膽固醇間不易形成致密結合,且原花青素對熱不穩定,因此成膜時水浴溫度不宜超過50℃。制備原花青素脂質體,固定其他組分的比例不變,僅改變水浴的溫度,包埋率隨溫度的變化情況見圖5。
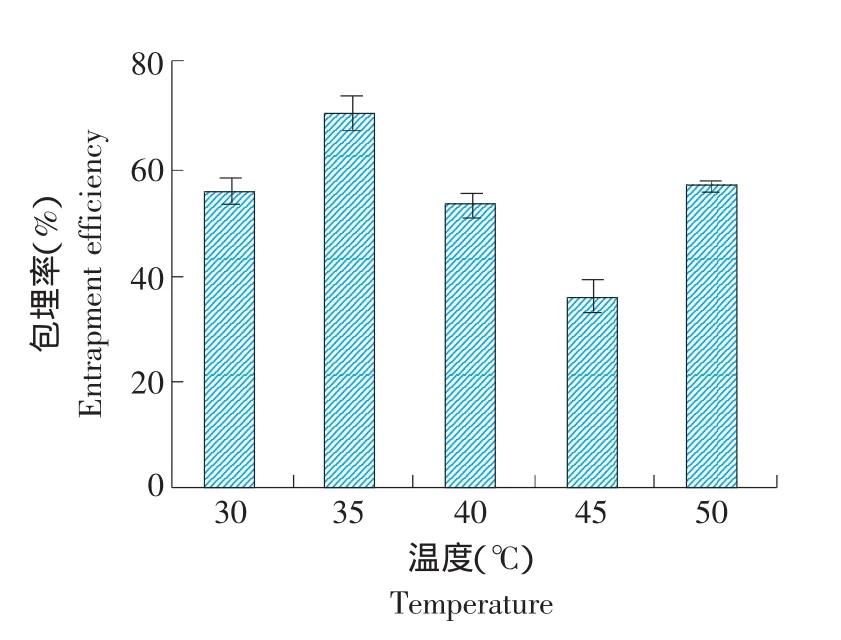
圖5 溫度對包埋率的影響Fig.5 Effect of the temperature on entrapment efficiency
在制備過程中,溫度對包埋率有較大影響,溫度過高時,脂質體流動性變大,膜內包封的物質易于泄露;而溫度過低時,有機溶劑的揮發速度變慢,影響脂質體的形成。由圖5可知,35℃時原花青素脂質體的包埋率最高,而50℃時的包埋率較45℃時高,原因可能是原花青素的熱穩定性差,導致其熱降解,故所得包埋率較高。
2.6 正交設計確定原花青素脂質體配方
根據初步試驗分析,影響原花青素脂質體包埋率的主要因素有膽固醇與卵磷脂物質的量比、原花青素與卵磷脂質量比、水溶液與有機溶劑體積比、溫度。因此用L9(34)正交試驗設計法進行試驗,確定原花青素脂質體的配方比例。

表1 正交試驗設計Table 1 Orthogonal design
根據單因素試驗結果,按照表1正交試驗設計確定的配方,以原花青素的包埋率為指標,確定最佳配方,結果見表2。
由表2的K值分析可以看出,4個因素對包埋率的影響順序為A>B>D>C,即膽固醇與卵磷脂比>原花青素與卵磷脂比>溫度>水溶液與有機溶劑比。該正交實驗的最優工藝條件為A2B2C3D2,但是此條件所得出的包埋率不如試驗5的包埋率高。因此最佳工藝條件是A2B2C3D1,即膽固醇與卵磷脂的物質的量比為1:2,原花青素與卵磷脂的質量比為1:40,水溶液與有機溶劑比為1:4,溫度為30℃,以此工藝條件制備三次原花青素脂質體,計算出包埋率的平均值為88.89%±2.5%。方差分析表明(見表3),在本試驗所選取的因素和水平下,膽固醇與卵磷脂比對包埋率有顯著影響,是影響脂質體包埋率最主要的因素。
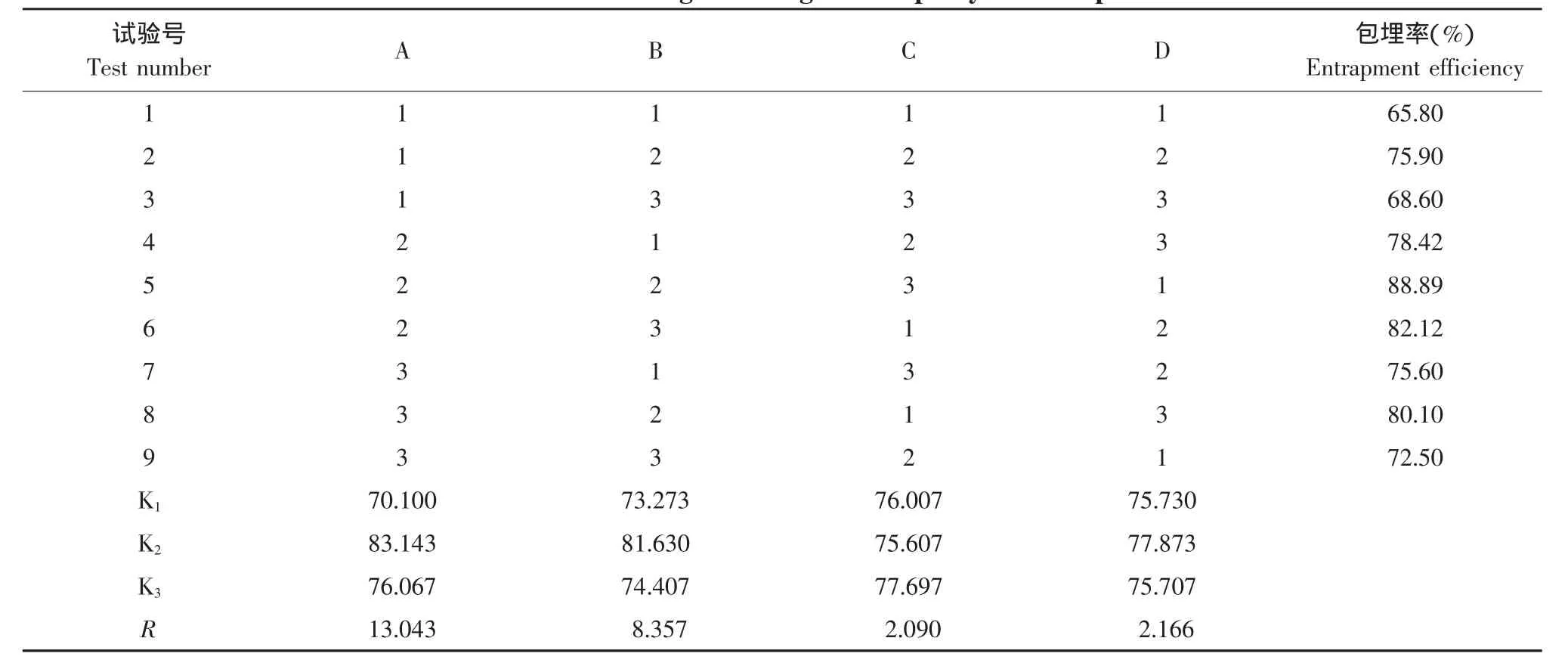
表2 原花青素脂質體正交設計結果Table 2 Results of orthogonal design about procyanidine liposomes
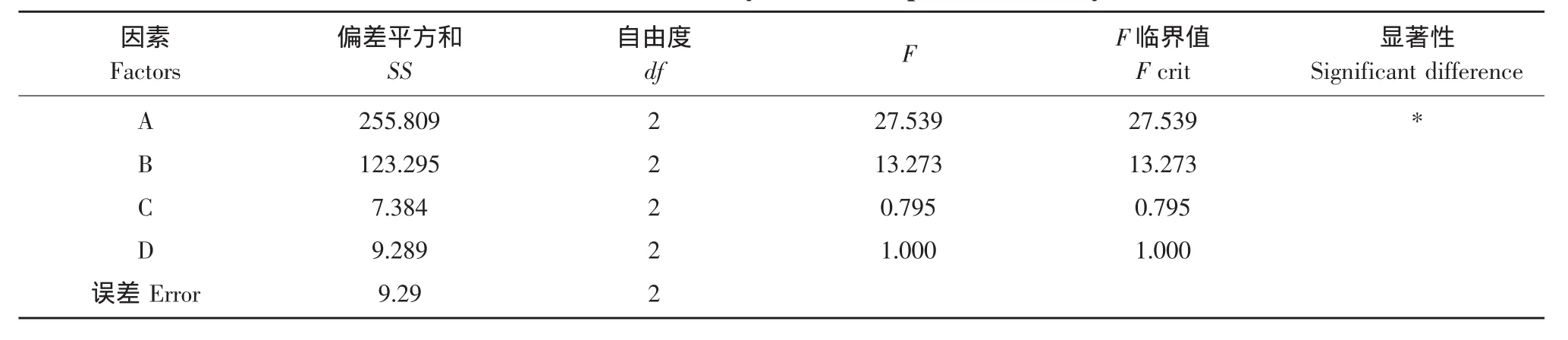
表3 包埋率方差分析Table 3 Variance analysis for entrapment efficiency
2.7 脂質體顯微形態觀察
對脂質體進行負染色后,在透射電子顯微鏡下觀察脂質體微觀結構(見圖6),呈球狀或近似球狀的小囊泡,分布均勻且顆粒間彼此分散。

圖6 逆相蒸發法制備的原花青素脂質體的復染色電鏡Fig.6 Electron microscope graph of procyanidine liposomes prepared by reverse evaporation
2.8 脂質體粒徑的測定
經DelsaNano C粒度分析儀測定原花青素脂質體的粒徑,脂質體的粒徑分布在185 nm~1.29 μm之間,平均粒徑為715.9 nm。
2.9 脂質體Zeta電位的測定
經DelsaNano C電位儀測定原花青素脂質體的Zeta電位。一般情況下,Zeta電位的絕對值越大,說明膠體體系越穩定,當電位>60 mV時體系穩定,電位30~60 mV時體系比較穩定,電位<30 mV時體系不穩定。因此,Zeta電位是衡量膠體穩定性的一個重要參數。測得的Zeta電位為37.04 mV,因此制備的原花青素脂質體混懸液是比較穩定的體系。
3 討論與結論
本研究在單因素試驗基礎上,經正交設計試驗分析得出最佳工藝組合為A2B2C3D2,各因素對包埋率的影響順序為A>B>D>C,即膽固醇與卵磷脂比>原花青素與卵磷脂比>溫度>水溶液與有機溶劑比,但在此條件下所得出的包埋率較A2B2C3D1所得到的結果低,分析其原因,可能是水浴溫度高時脂質的成膜速度快,卵磷脂和膽固醇間不易形成致密結合,從而影響脂質體的包埋率。考慮到本試驗的目的是制備包埋率高的原花青素脂質體,故確定處方工藝組合為A2B2C3D1,即膽固醇與卵磷脂的物質的量比為1:2、原花青素與卵磷脂的質量比為1:40、水溶液與有機溶劑比為1:4、溫度為30℃。在此條件下,原花青素脂質體的包埋率為88.89%±2.5%。
脂質體的包埋體積和脂質體的粒徑呈線性關系,主要取決于包埋的水相體積[16]。粒徑越小包埋的水相體積越小,會降低脂質體的包埋率。本研究中制備的脂質體為大單室脂質體,其具有較大的水相容積,平均粒徑為715.9 nm,包埋率較高。Zeta電位測定表明原花青素脂質體電位處于30~60 mV之間,是比較穩定的體系。此種制備方法操作方便、設備簡單,適合工業化生產。
[1] 張冰若,勞業興,蘇薇薇.原花青素的研究現狀及開發前景[J].中藥材,2003,26(12):905-908.
[2] 國植,徐莉.原花青素:具有廣闊發展前景的植物藥[J].國外醫學植物藥分冊,1996(11):196-201.
[3] Merfort HeilmannJ,WeissM.Radical scaven geactivity of three flavonoid metabolites stdied by inhibition of chemiluminesscence in human PMNs[J].Planta Medica,1996,62(4):289-292.
[4] Packer L,Rimbach G,Virgili F.Antioxidant activity and biologic properties of a procyanidin-rich extract from pine(Pinus maritma)bark,pycnogenol[J].Free Radical Biology and Medicine,1999,27(5-6):704-724.
[5] 叢紅群,成漢義,鐘進義.葡多酚對N-亞硝基化合物誘變性的抑制作用[J].癌變·畸變·突變,2004,16(1):30-33.
[6] Bagchi D,Garg A,Krohn R L,et al.Oxygen free radical scavenging abilities of VC and VE,and a grape seed proanthocyanidins extract in vitro[J].Research Communications in Molecular Pathology and Pharmacology,1997,95(2):179-189
[7] Guy R C E,Home A W.Food structure-its creation and evalution[M].London:Butterworths,1988:331-349.
[8] 韓菊,魏福祥,麗萍,等.葡萄籽中低聚原花色素的性能研究[J].食品科學,2003,24(2):36-38.
[9] 鄭燕升,莫倩,廖政達.野生毛葡萄籽原花青素的穩定性研究[J].安徽農業科學,2008,36(13):5259-5260.
[10] 張琦,孟憲軍,孫希云,等.葡萄籽中原花青素的穩定性研究[J].沈陽農業大學學報,2006,37(2):232-234.
[11] Kobdo K,Uchild R,Tokutake S,et al.Polymeric grape seed procyanidins,but not monomeric catechins and oligomeric procyanidins,impair degranulation and membrane ruffling in RBL-2H3 cells[J].Bioorganic and Medicinal Chemistry,2006,14:641-649.
[12] 唐培宇,劉寧.與磁性納米粒結合的抗腫瘤方法[J].東北農業大學學報,2007,38(3):420-423.
[13] Szoka F J R.Procedure for preparation of liposomes with large internal aqueous space and high capture by reverse-phase evaporation[J].Proceedings of the National Academy of Sciences,1978,75(9):4194-4198.
[14] 任文霞,李建科.茶多酚脂質體的制備[J].食品工業科技,2008(11):186-191.
[15] 周威.脂質體不同制備方法對其包裹率的影響[J].武漢工業學院學報,2001(2):28-29.
[16] Bamadas-Rodriguez R,Sabes M.Factors involved in the production of liposomes with a high-pressure homogenizer[J].International Journal of Pharmaceutics,2001,213(1/2):175-186.
