再談什么是活化能
——Arrhenius活化能的定義、解釋、以及容易混淆的物理量
2010-07-02 00:34:38羅渝然俞書勤張祖德姚天揚高盤良
大學化學 2010年3期
關鍵詞:理論
羅渝然俞書勤張祖德姚天揚高盤良
(1中國科學技術大學化學與材料科學學院 安徽合肥230026;2南京大學化學化工學院 江蘇南京210093;3北京大學化學與分子工程學院 北京100871)
知識介紹
再談什么是活化能
——Arrhenius活化能的定義、解釋、以及容易混淆的物理量
羅渝然1俞書勤1張祖德1姚天揚2高盤良3
(1中國科學技術大學化學與材料科學學院 安徽合肥230026;2南京大學化學化工學院 江蘇南京210093;3北京大學化學與分子工程學院 北京100871)
介紹IUPAC(1996)推薦的活化能定義,它源于反應速率常數與溫度關系的Arrhenius圖。對基元反應,活化能的Tolman解釋最準確;對總包反應,活化能僅是表觀量。以基元反應D+ H2為例,強調了活化能與許多相似物理量的區別。在較寬溫度范圍內,該Arrhenius圖是彎曲的,這表示反應活化能隨溫度而變化。討論為什么少數基元反應的活化能可能是負值甚至是零。
中學、大學與研究生用的化學參考教材中,活化能是出現頻率很高的科學術語之一。但是,什么是活化能?不少國內大學教材和教學參考資料仍在使用不準確的文字。下面,我們將再次介紹有關Arrhenius活化能的定義、解釋、以及很容易與活化能混淆的若干概念。此外,還討論了非Arrhenius行為和負溫度效應等。
1 簡單的Arrhenius方程與活化能的準確定義
溫度對許多化學反應速率有非常大的影響。1889年,瑞典科學家S.A.Arrhenius在研究蔗糖水解的速率與溫度的關系時,受van't Hoff類似工作的啟發,用速率常數k的自然對數ln k對溫度的倒數1/T作圖,得到一條直線。在科學文獻上,ln k(或log k)對1/T作圖,簡稱Arrhenius圖。Arrhenius圖上的線性關系可表示為:

引入氣體常數R,可將這樣的線性關系改寫為:

或

上面的經驗表達式(2)和(3),都稱Arrhenius公式,它包括2個重要的經驗參數A和Ea,這2個經驗參數通稱Arrhenius參數。其中參數A稱為A因子(或指前因子),與速率常數k有相同單位,它來自Arrhenius圖上的截距。Arrhenius把由圖上斜率導出的能量因子Ea稱為“活化熱”,后來被科學文獻改稱為Arrhenius活化能。按照IUPAC(1996)推薦的觀點[1],活化能Ea的準確定義是Arrhenius圖上該直(曲)線在溫度T下的斜率[1]:

從活化能的定義(式(4))看,反應溫度T升高,速率常數k相應增加,活化能Ea必為正值。我們稱它為正溫度效應。絕大多數化學反應呈正溫度效應。反之,稱為負溫度效應,對應于負活化能(見本文第5節和圖3)。實驗發現,在極少數情況下,某一很小的溫度范圍內,溫度升高或降低,反應速率常數不變化,這對應于零活化能(見圖3)。
基元反應的Arrhenius圖中的速率常數k可從實驗獲得,也可按反應速率理論計算。因此,既可利用測量的k(T)數據得到基元反應的活化能,也可按反應速率理論計算的k(T)數據得到活化能(見本文第3節)。目前,國內某些大學教材和教學討論文章還未接受IUPAC (1996)推薦的觀點,賦予化學反應活化能以另外的定義,錯誤地認為從量子理論或某些理論可直接計算出活化能。
2 基元化學反應的Arrhenius活化能的最合理解釋
Arrhenius經驗式的提出,標志了定量的化學反應動力學誕生。100多年來,大量的實驗發現,Arrhenius經驗公式有很強的預測能力。從化學反應的3個層次看[2-7],在宏觀(總包和基元)層次上研究化學反應,經常可繪出Arrhenius圖,采用Arrhenius活化能的概念,如表1所示。這是因為,只要化學反應速率受溫度影響,就可作Arrhenius圖,確定某溫度T下的斜率,即活化能Ea值。
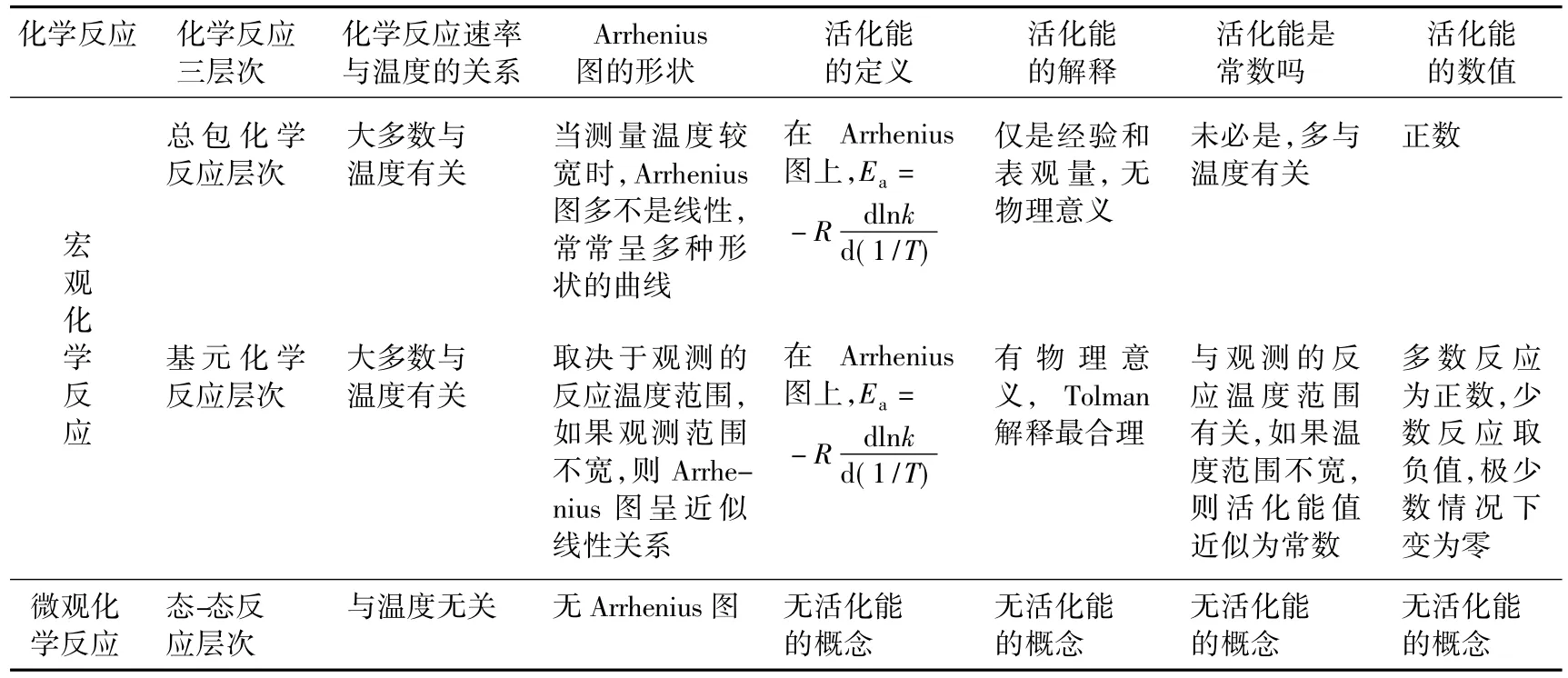
表1 化學反應三層次與活化能概念的適宜范圍
活化能Ea位于Arrhenius公式中的指數項,因此活化能數值的改變,對化學反應速率常數有極大的影響,遠比A因子的影響強烈。例如,常溫條件下,活化能每改變5.7kJ/mol,反應速率常數將變化一個數量級。
對于如何理解Arrhenius活化能以及它有什么深入的物理意義,是100多年來人們一直感興趣的科學問題。科學家的長期努力,已促進了若干化學速率理論的誕生。隨著科學的進步,對基元反應活化能的理解歷經了如下主要觀點的變遷[8]:
①活化能是由非活化分子轉變為活化分子的能量(Arrhenius);
②具有完成化學反應最小的、必須的能量(Lewis);

③在反應溫度T時,全部活化分子的平均能量與全部反應物分子平均能量之差(Tolman),即:這里,對稱符號<>表示對其中的物理量取統計平均。以上3種解釋中,Tolman從統計力學提供的解釋最準確、最合理[1-8]。雖然Tolman的論述發表在20世紀20年代,但長期沒有得到人們的足夠關注。到20世紀70年代,隨著分子反應動態學(或微觀化學動力學)的興起和發展,國內外化學教材開始廣泛傳播Tolman的統計觀點。Tolman解釋有最清晰的物理意義,是因為人們觀測到的基元化學反應都是一大群分子統計平均的結果,科學家還不能測量僅僅一個分子的行為。
正如表1強調的,在總包化學反應的層次上(如燃燒和爆炸、熱裂解、多相催化、酶催化、有機物氧化或鹵化、聚合反應等),當測量溫度比較寬時,Arrhenius圖多不是線性,常常呈多種形狀的曲線[4]。按活化能定義(式(4)),它必然不是一常量,隨溫度變化。并且,這時候得到的活化能數值僅是經驗和表觀量,沒有物理意義。這樣情況下,如果把表觀活化能叫E因子,也許會減少混淆。同時,表1還指出,Arrhenius圖與活化能的概念不適合態-態反應。因為,在態-態反應層次上,沒有熱力學溫度T的概念[8]。
大學教材在介紹化學反應的3個不同層次之后,不僅可以理解Arrhenius方程的局限性(A和Ea未必是常數,取決于反應溫度范圍),而且可以認識質量作用定律的局限性(不能用于總包反應,僅適合基元和態-態反應)。
3 Arrhenius活化能與反應能(位)壘、Eying理論的活化焓、活化內能、態-態反應的臨界(閾)能等的嚴格區別
在化學文獻中,反應勢能面上的能(位)壘、Eying理論的活化焓、活化內能、態-態反應的臨界(閾)能等是經常與Arrhenius活化能混淆的物理量。Pacey[9]用圖示(圖1)說明了這些物理量之間的區別。
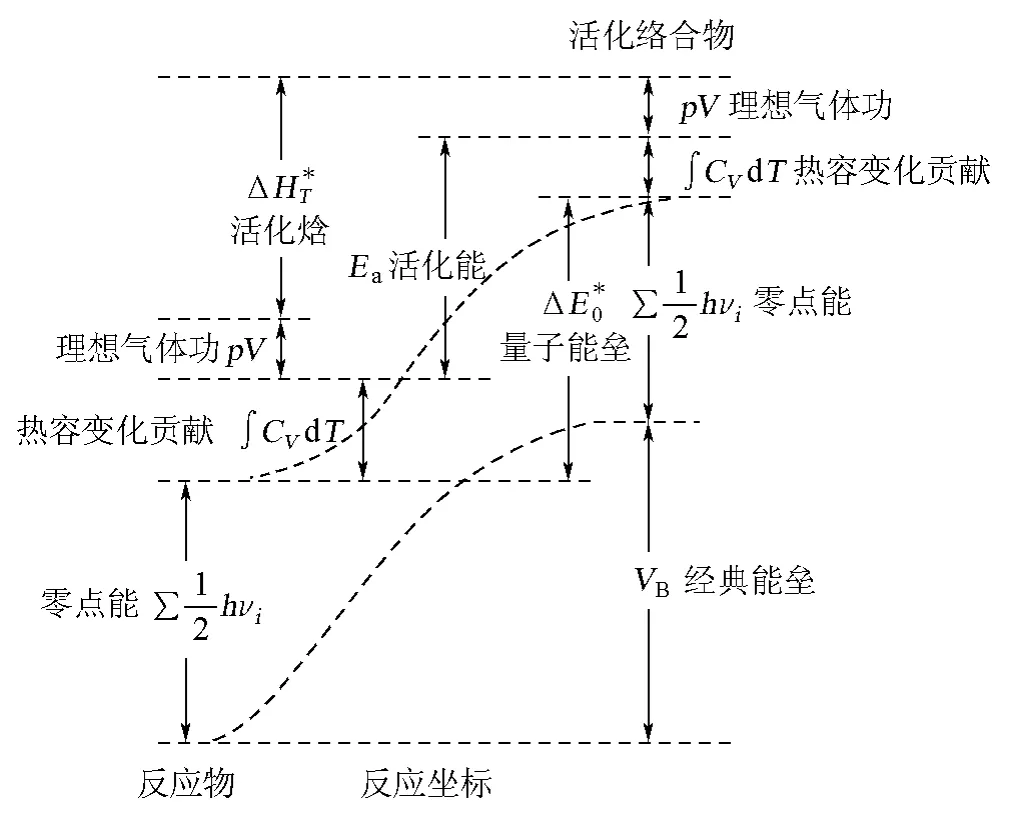
圖1 幾個容易與Arrhenius活化能混淆的物理量[9]
只有嚴格區分這些相似的物理量,化學反應動力學的研究及其教學才是嚴格和清楚的。表2給出了全部7個物理量的定義、特點和取值范圍。在閱讀圖1時,要注意哪些量是0 K時的物理量,哪些量與熱容和溫度的變化有關。在理解圖1和表2時,特別要注意4點:
①經典能壘和量子能壘之間有區別。從經典能壘到量子能壘,要對反應物和活化絡合物分子作零點能修正。量子化學計算出的勢能面和能壘高度僅是0K時的值。在0K時分子沒有平動能,沒有碰撞,沒有化學反應,勢能面上的能壘不等同于實際化學反應的活化能。
②活化焓與活化內能之間有區別。對1mol理想氣體,其差異是氣體做的功pV=RT(圖1),在室溫條件下,它等于2.5kJ/mol。
③對于室溫(或另外溫度T)下的化學反應,在考慮0K時的量子修正之后,還要分別對反應物和活化絡合物作從0K到實際溫度T的熱容變化的修正項(∫CVd T)。
④閾(臨界)能是實驗(如交叉分子束技術)可觀測到的微觀量,是分子水平上的量,與熱力學溫度T無關。閾能是發生反應性碰撞時分子必須具有的最低能量,它表明反應途徑上有能壘。對于隧道效應較明顯的化學反應,閾能為零。
以基元反應D+H2(DH+H為例,Pacey[9]給出了能壘(經典或量子)、閾能(理論或實驗)、過渡態理論中的能量參數(活化焓或活化內能)等與該反應活化能之間在數值上的區別。它們列在表2的最后一行,能量單位是kJ/mol。
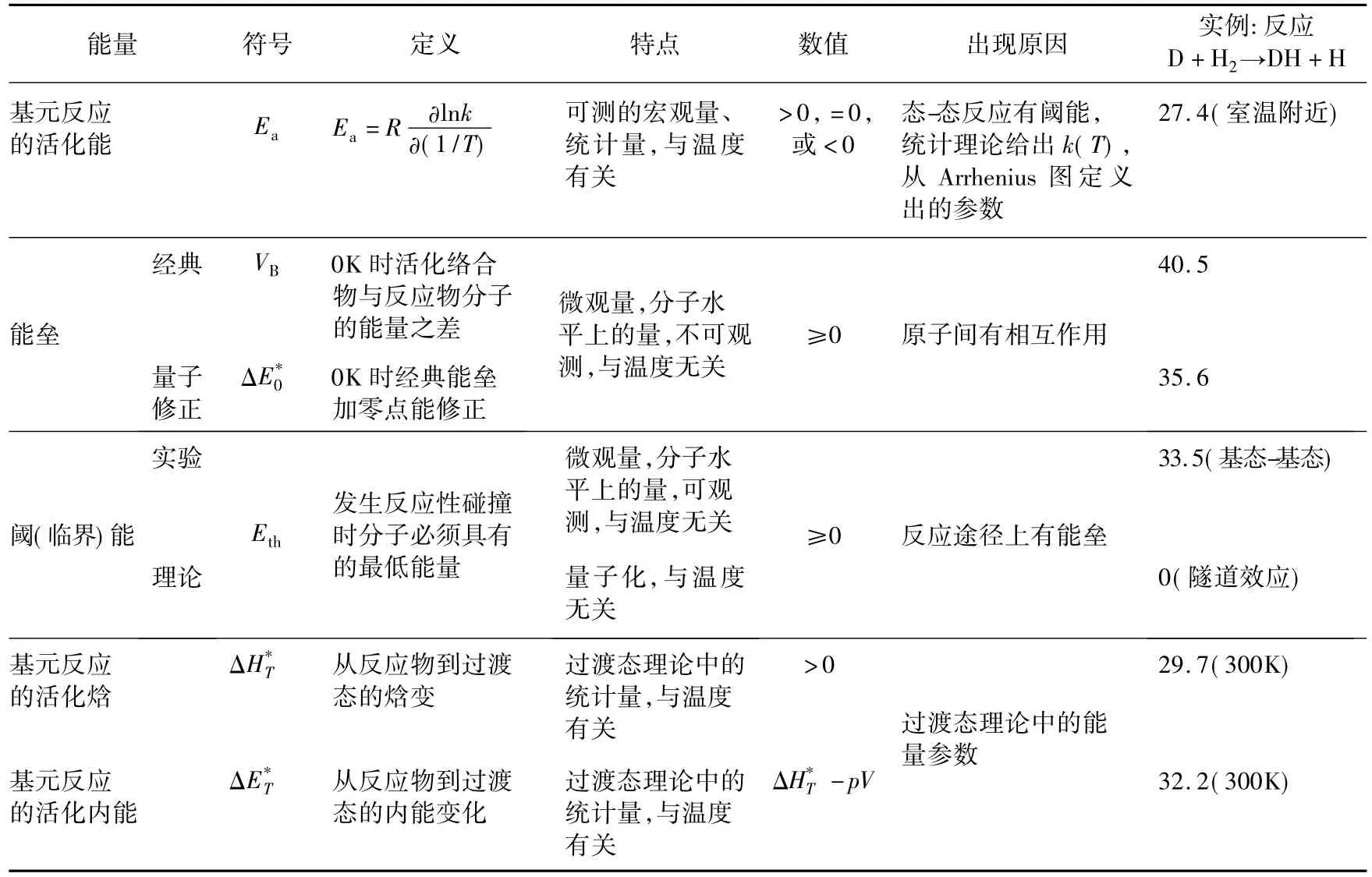
表2 幾種容易與Arrhenius活化能相混淆的物理量(能量單位k J/mol)
反應勢能面上的能(位)壘、Eying理論的活化焓、活化內能、態-態反應的臨界(閾)能等與Arrhenius活化能有本質上的區別,但也有聯系。一般地說,通過統計理論,利用微觀的能壘和閾能等參數,可推導出該基元反應在不同溫度下的速率常數k(T),作Arrhenius圖,就可估算出活化能等Arrhenius參數。過程如下:

在有關理論(例如過渡態理論)的骨架下,在計算了該基元反應不同溫度下的速率常數k(T)后,作Arrhenius圖,也可得到活化能等Arrhenius參數。

如果讀者對上述理論有興趣,可閱讀參考文獻[2-4]。針對目前國內大學教材中的現況,下面再補充3點:
①利用過渡態理論可直接計算活化能嗎?目前已發表了許多基元化學反應速率理論,至少已有20多種。甚至過渡態理論也有10多種,它們早已有別于20世紀30年代的傳統過渡態理論。所有的反應速率理論都是建立在某一勢能面上,然后用不同的統計方法,計算基元反應在不同溫度下的速率常數k(T)。只有作Arrhenius圖后,才能確定該基元反應的活化能。換句話說,任何反應速率理論(包括種種過渡態理論)都不可能直接計算出活化能。
②實驗活化能與理論活化能有什么區別?按IUPAC(1996)推薦的觀點[1],活化能的正確理解僅來自Arrhenius圖的斜率(即式(4)),如果Arrhenius圖中的k(T)來自實驗,就可稱它為“實驗活化能”;如果Arrhenius圖中的k(T)來自反應速率理論計算,可稱它為“理論(或計算)活化能”。此外,不少化學文獻借助于分子模型和幾條理論假設,避開研究反應速率常數k和溫度T的關系,力圖直接取得基元反應活化能的數值。這樣得到的活化能數值,只能叫預測的(predicted)或估算的(estimated)活化能。換句話說,預測(或估算)的活化能與理論(或計算)活化能是兩回事,在邏輯上和概念上不可混淆。
③表觀活化能和基元反應活化能有什么區別?因為我們既可觀測總包反應速率常數k隨溫度T的變化,還可測量基元反應的k(T)關系,故對這兩種不同層次的化學反應,都可作ln k (T)-1/T圖。從總包反應實驗數據k(T)得到的活化能,就是“表觀活化能”,它是一個表觀量,沒有物理意義。表2和式(5)指出,基元反應的活化能才具有明確的物理意義。
4 改進的Arrhenius方程、活化能的數值隨溫度變化
在Arrhenius研究蔗糖水解動力學的年代,測量的溫度范圍比較小。故在Arrhenius圖上,二元線性回歸方程(3)很成功,A因子與活化能Ea被認為是一常數。但是,如果觀測的溫度范圍比較大時,通常三元回歸方程才能提供較好的擬合:

或

式(6)和(7)都稱為改進的Arrhenius方程。這里,A,A'和E'都是與溫度無關的常數,Tref是參考溫度,常設Tref=298.15K;n可以是正數、負數、整數或分數。注意,式(6)或(7)中的物理量E'不能理解為活化能,稱它能量因子或E因子更妥當。把式(6)或(7)代入式(4),可得:

按式(8),Arrhenius活化能Ea與溫度有關。下面舉一例子。
在167~1980K的溫度范圍,Michael等[10]用閃光光解和激波技術觀測了基元反應D+H2→DH+H的速率常數。他們在Arrhenius圖上(圖2)發現觀測點可用改進的Arrhenius方程描述:
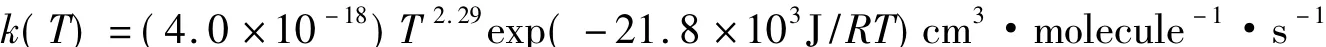
在NIST化學動力學數據庫里(http://kinetics.nist.gov/kinetics),Michael等的結果被改寫為:
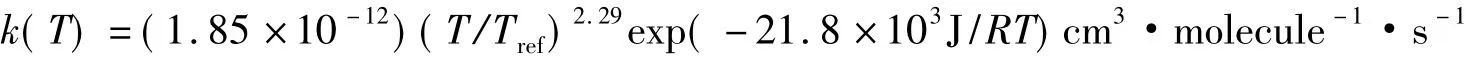
NIST數據庫也用簡單的Arrhenius方程回歸了Michael等的結果,如圖2所示。在圖2中,彎曲線是改進的Arrhenius方程的擬合,直線是簡單的Arrhenius方程的擬合。表3對比了這兩種擬合結果。從均方根偏差看,用改進的Arrhenius方程擬合提供了很好的結果,其均方根偏差幾乎為零。在科學文獻中,把這樣的關系叫非Arrhenius行為。大多數基元化學反應都呈現非Arrhenius行為。
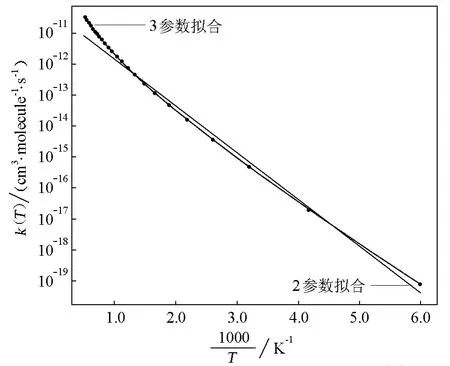
圖2 反應D+H2→DH+H的Arrhenius圖[11]

表3 基元反應D+H2→DH+H速率常數的擬合(T=167~1980K)
Michael等[11]最近發現,在167~2112K溫度范圍內,為了得到最好的擬合,建議對于基元反應D+H2采用如下多項式:
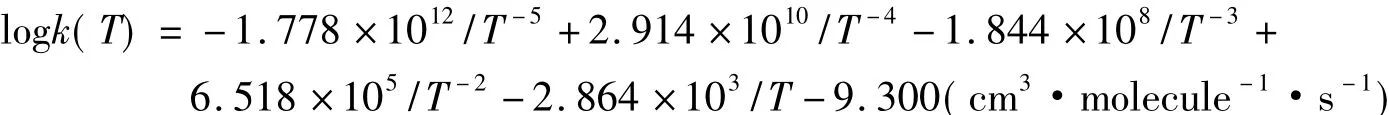
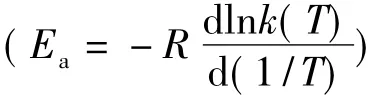
在某些情況下,改建的Arrhenius方程可能退化為:

這時,反應活化能可簡單地估算:

5 基元化學反應的負溫度效應與負活化能
Smith等[12]歸納了許多OH的奪H反應(OH+RH→H2O+R)的速率常數,他們發現,某些反應有負溫度行為,即溫度升高,速率常數降低,如圖3所示。負溫度效應已被許多科學家觀測到。顯然,負溫度行為對應于負活化能。圖3還表示,對于基元反應

在高溫區間(>300K),呈正活化能;在低溫區間(<290K)時,呈負活化能;在一很小的溫度區間(點),該反應的活化能是0(零活化能)。
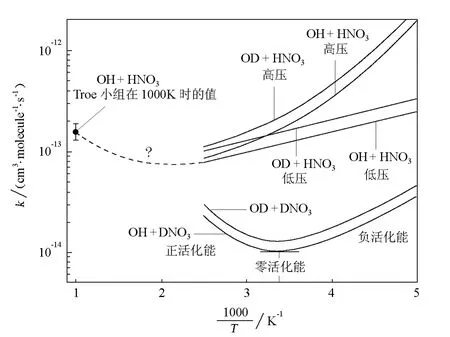
圖3 某些OH奪H反應的Arrhenius圖[12]
Smith等認為[12],該基元反應經歷了氫鍵(締合的中間)絡合物(圖4)。
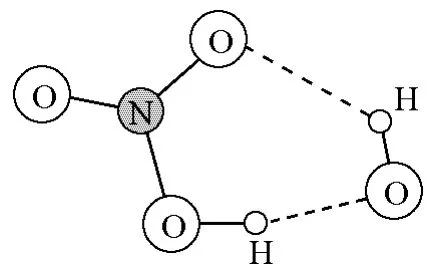
圖4 OH奪H反應形成的氫鍵絡合物
該氫鍵絡合物的能量比反應分子的能量低,在勢能面上出現一個“勢阱”(如圖5)。有負溫度行為的許多基元反應的勢能面都有類似的“勢阱”。在態-態反應層次(表1),它們對應于長壽命絡合物[4,8],不同于短壽命絡合物的動力學特征。
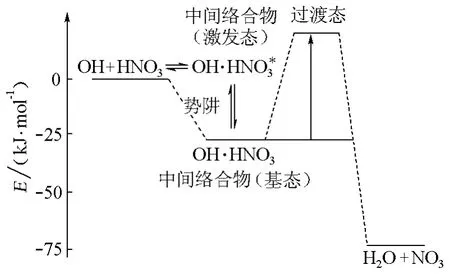
圖5 OH+HNO3的反應機理與氫鍵(締合的中間)絡合物[12]
基元化學反應的負溫度效應與負活化能的研究,使我們對化學反應的多樣性和極其復雜性,有了更深入、更全面的認識。
6 建議和結束語
①大學教材應當與時俱進,接受IUPAC(1996)推薦的活化能定義,放棄其他陳舊的說法。
②物理化學教材應當嚴格區別Arrhenius活化能與反應能(位)壘、Eying理論的活化焓、活化內能、態-態反應的臨界(閾)能等許多相似物理量。
③大學教材應當指出,Arrhenius圖既可用2參數擬合(Arrhenius方程),也可用3參數擬合(改進的Arrhenius方程)。當實驗溫度的范圍較寬時,改進Arrhenius方程擬合的均方根偏差最小。
④在物理化學教材中應當闡明,為什么基元反應的活化能不一定是常量,為什么少數基元反應的活化能可能是負值甚至是0。
[1] Laidler K J.Pure&Appl Chem,1996,68:149
[2] 俞書勤.微觀化學反應.合肥:安徽科學技術出版社,1985
[3] 韓德剛,高盤良.化學動力學基礎.北京:北京大學出版社,1987
[4] 趙學莊,羅渝然,臧雅茹,等.化學反應動力學原理(下).北京:高等教育出版社,1990
[5] 胡英.物理化學參考.北京:高等教育出版社,2003
[6] 范康年.物理化學.第2版.北京:高等教育出版社,2005
[7] 印永嘉,姚天揚.化學原理.北京:高等教育出版社,2006
[8] 羅渝然.化學通報,1981(4):50
[9] Pacey PD.JChem Educ,1981,58:612
[10] Michael JV,Fisher JR.JPhys Chem,1990,94:3318
[11] Michael JV,Su M C,Sutherland JW.JPhys Chem A,2004,108:432
[12] Smith IW M,Ravishankara A R.JPhys Chem A,2002,106:4798
猜你喜歡
當代陜西(2022年5期)2022-04-19 12:10:18
新世紀智能(數學備考)(2021年9期)2021-11-24 01:14:28
湘潮(上半月)(2021年4期)2021-07-20 08:05:28
汕頭大學學報(自然科學版)(2020年4期)2020-12-14 07:05:00
讀與寫·教育教學版(2017年10期)2017-11-10 22:28:57
大電機技術(2017年3期)2017-06-05 09:36:02
廣州大學學報(社會科學版)(2016年1期)2016-06-24 09:46:02
區域經濟評論(2016年2期)2016-05-17 05:06:43
學習月刊(2015年21期)2015-07-11 01:51:44
社會生活探索(2013年0期)2013-10-24 03:44:40
