汽車三效催化劑失活研究的進展
2010-01-11 08:02:32卞龍春盛世才隨偉張保才
浙江化工 2010年12期
關鍵詞:催化劑
卞龍春 盛世才 隨偉 張保才
(寧波科森凈化器制造有限公司,浙江 寧波 315145)
0 前言
隨著全球汽車數量的急劇增加和排放法規的日益嚴格,尾氣排放問題越來越受到重視,同時對汽車三效催化劑的耐久性能提出了更高的要求。早在2001年,我國就對機動車尾氣催化劑耐久性提出相關要求,即:陶瓷蜂窩載體催化劑>5萬km(在用車);陶瓷蜂窩載體催化劑>8萬km(新車);金屬載體催化劑>10萬km。汽車三效催化劑的使用壽命是衡量催化劑性能的重要指標,催化劑的壽命與它的行駛條件、環境及工況有關,且影響催化劑工作的因素很多,汽車在行駛的過程中,汽車三效催化劑要受到排出尾氣的沖刷,要經歷溫差達幾百度的熱循環、熱疲勞和熱沖擊等十分惡劣條件的考驗,從而導致催化劑的失活,三效催化劑失活的現象為催化劑表面的活性位減少,引起催化活性的下降。三效催化劑的失活主要可分為三種類型:機械失活、熱力學失活及化學失活。
1 汽車尾氣催化劑機械失活
機械失活主要是汽車在行駛的過程中尾部的催化轉換器受到沖擊,壓輾等機械變化而引起的催化劑的損壞。早期的顆粒狀載體由于強度低、易破碎等特點最易發生機械失活,對于目前使用最多的汽車催化劑載體陶瓷蜂窩狀載體而言,這也是一種正常的失活現象,而金屬蜂窩載體具有機械強度高,防震性能好等優點很難發生機械失活。但金屬載體材料的成型工藝較復雜,與活性表面層的附著力較弱及成本較高還未得到普遍應用。因此目前催化劑的失活研究主要集中在陶瓷峰窩狀載體的催化劑上。
2 汽車三效催化劑的熱失活
將催化劑安裝靠近汽車發動機附近的緊密耦合技術,被認為是最有效降低冷起動排放的方法。然而,緊密耦合催化劑要承受比底盤催化劑高很多的溫度,加上反應的劇烈放熱,瞬間溫度可達1000℃以上,從而引起了催化劑的結構和性能的變化,是典型的汽車尾氣催化劑熱失活,主要現象包括涂層基體γ-Al2O3的相變和活性組分貴金屬顆粒的燒結。
2.1 涂層基體γ-Al2O3老化
氧化鋁涂層的相變和燒結是機動車尾氣催化劑高溫老化的一個重要原因。γ-Al2O3的比表面積越大,貴金屬顆粒在其表面分散的程度就越大,活性表面越大。由于γ-Al2O3為介穩態,在高溫下易發生相變和燒結,向熱力學上穩定的α相和大顆粒化轉變,通常在800℃以下氧化鋁以γ-Al2O3形式存在,1100℃下其晶相轉變為α-Al2O3。目前活性氧化鋁涂層的燒結和相變機理可用羥基脫水縮合來解釋,Burtin[3]以Al2VMO(3-v/2)(OH)vVo(1-v/2)(VM表示陽離子空穴,Vo表示O2-空穴)來表示氧化鋁,v=0或2分別表示α-Al2O3(無羥基)和薄水鋁石(無O2-空穴)。Al2O3相變的過程可以解釋為脫羥基的過程:
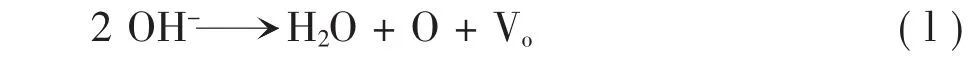
只要溫度足夠高,脫羥基過程就會不斷的進行。伴隨脫羥基過程中O2-空穴濃度隨之增加至結構完全脫離羥基,氧化鋁內部的因缺陷產生的陽離子空穴和脫羥基產生的陰離子空穴會相互中和,導致氧化鋁晶格完全破壞,生成α-Al2O3晶相。

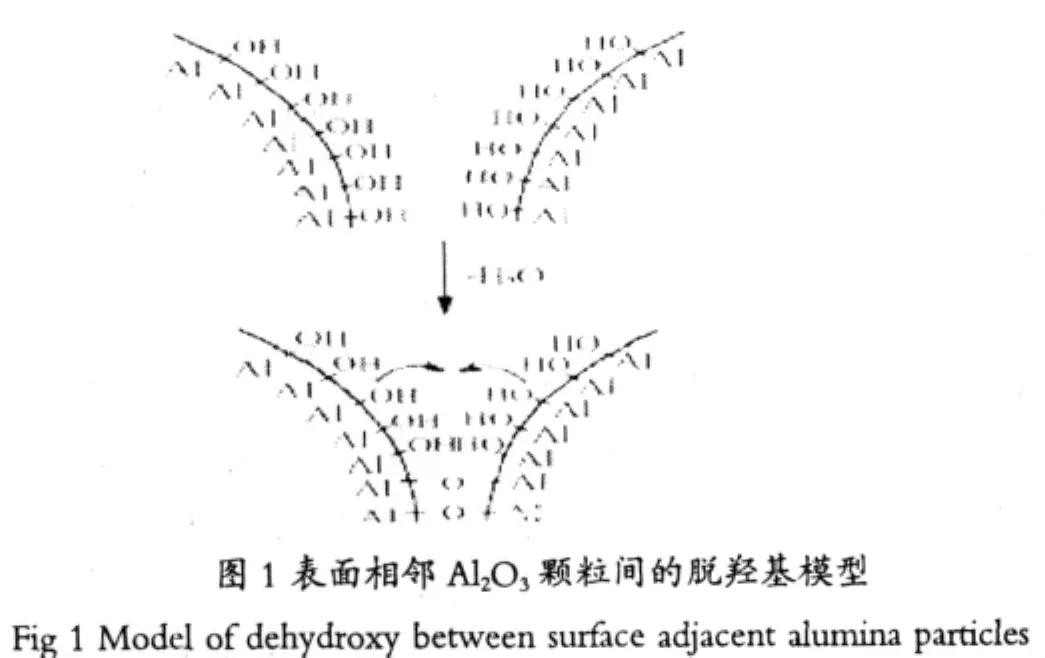
Burtin的燒結模型指出,α-Al2O3的形成包括兩個過程即成核和核生長。Johnson[7]研究了Al2O3在焙燒時的燒結問題,提出了相鄰氧化鋁顆粒間的脫羥基模型(圖1)。依據模型可知,氧化鋁顆粒的生長是通過顆粒間相互接觸部分的羥基基團脫水實現的,隨著水分子的不斷脫除Al-O-Al鍵生成,從而導致氧化鋁的表面積衰減。
2.2 貴金屬的老化
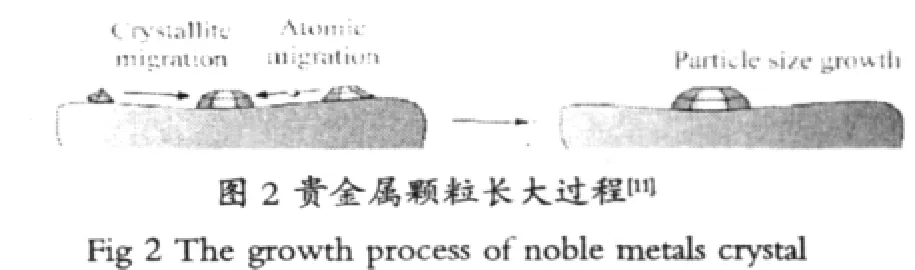
貴金屬顆粒的燒結是汽車三效催化劑熱老化的另一主要現象,Beck等[5]研究了三效催化劑的催化活性與貴金屬Pt、Pd及Rh的顆粒之間的關系,結果表明,兩者呈線性關系,當溫度越低貴金屬催化活性越高。因此抑制貴金屬顆粒變大非常重要。通過掃描電鏡分析表明,貴金屬顆粒是以納米級的超細微粒分散在氧化鋁中時,由于貴金屬以納米級顆粒分布,故具有很高的催化活性。催化劑經高溫老化后,比表面收縮和晶粒的長大,貴金屬顆粒補包裹,使得H2與亞晶格氧的接觸機會減小,抑制了還原反應發生:同時,高溫老化使晶化度增強,降低了氧的可移動性[6]。大量實驗表明,貴金屬和貴金屬氧化物更易發生燒結,由于它們均以細小晶粒存在,溫度過高時它們與載體發生反應,從而使得反應活性中心大幅度減小。貴金屬和貴金屬氧化物催化劑活性組分燒結機理也可能存在兩個規律,即活性組分晶粒遷移和原子遷移[7]。
3 汽車三效催化劑的化學失活
催化劑的化學失活主要是化學中毒,可定義為由于雜質在活性位上的化學吸附而導致催化劑活性的下降,這些雜質主要來源于燃油和潤滑油中的添加劑。催化劑的中毒可分為可逆和不可逆的。
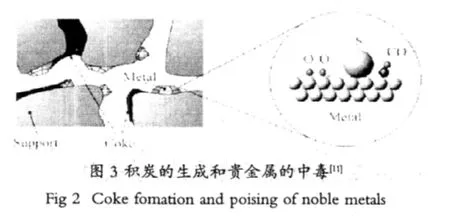
焦炭的生成是一種可逆的物理現象(圖3),涂層基體氧化鋁的孔結構中微孔所占的比例很高,因此在反應過程中由于焦炭的生成會堵塞微孔,不利于廢氣和熱量在氧化鋁孔結構中的流通,易導致催化劑失活和活性下降,是最常見的失活方式。這種可逆的失活可通過化學手段去除吸附在催化劑上的碳而恢復催化劑活性。
不可逆中毒類型包括Pb、S、P、Mn以及鹵化物中毒等。通常,鹵化物、Pb和Mn與燃料添加劑有關,而P、Zn和Ca則在潤滑油中使用,兩種燃料中都含有S。汽油中添加Pb是為了提高汽油的防爆性能,早期的汽油中鉛含量較高,易導致汽車催化劑的永久失活。鉛中毒引起的失活是不可逆的。Gandhi等[8]研究了Pb與貴金屬Pt、Pd、Rh的相互作用,在不同的催化劑載體上得到一層Pb與Pt、Pd、Rh的噴濺膜。結果發現,在700℃~800℃時,貴金屬活性組分中的Pd與Pb形成了固溶體,導致了催化劑的失活。
燃油中通常含有少量的S(<5×10-5),S能快速影響催化劑的性能和儲氧能力(OSC),硫中毒(見圖3)通常是在催化劑的表面形成無活性的化合物,也會使催化劑的形貌發生改變。由硫引起的汽車催化劑的快速中毒在某種程度上是不可逆轉的。Shaw等[9]指出中毒后的汽車催化劑在溫度低于650℃下是不可逆的,但是在高溫的時候其原始的催化活性可恢復,應當指出的是盡管凈化能力恢復了,但是儲氧能力不能恢復。在發動機燃燒過程中,硫氧化生成SO2和SO3,這些氧化物在低溫時(低于300℃)吸附在活性位上與氧化鋁生成硫酸鋁鹽,減少了涂層上活性位的比表面積。此外,空燃比也會影響硫的性能,貧燃時SO2儲存在CeO2上,富燃時,SO2和SO3都被還原成H2S,美國解決這個問題的方案是添加Ni到三效催化劑中。Ni作為S的清除劑(據稱在富燃時轉換成NiS),然后在貧燃時作為SO2釋放[10]。
為了減少鉛對催化劑的影響,通常往燃油中添加二氯化乙烯(EDC)和二溴化乙烯(EDB),EDC和EDB是機動車尾氣中鹵化物的來源。在高溫時由EDC、EDB分解得到的Cl和Br易揮發,不能直接引起催化劑的中毒。Shelef等[11]研究表明鉛凈化劑本身能夠抑制鉑、鈀催化劑的活性。鉛凈化劑引起的催化劑短暫性中毒主要和催化劑對EDC和EDB的相對吸附有關,在CO、HC催化氧化過程中,催化劑活性位上會發生競爭性吸附,鹵素物種相比于CO、HC更容易吸附在催化劑表面,從而引起催化劑短暫性中毒失活。
鹵化物、Pb和S的中毒現象逐漸減少,因為它們在汽油中含量很低而且催化劑配方也得到改進。更加嚴格的法規降低了排放限值,盡量減少催化劑失活顯得非常重要,目前的焦點主要集中在由Zn、Ca和P化合物引起的中毒,它們分別來自于石油酸中和,抗氧化劑和提高發動機耐久性的抗磨損劑。雙有機二硫代磷酸鋅(ZDDP)作為一種強效的抗磨損劑和抗氧化劑廣泛應用于潤滑油中。
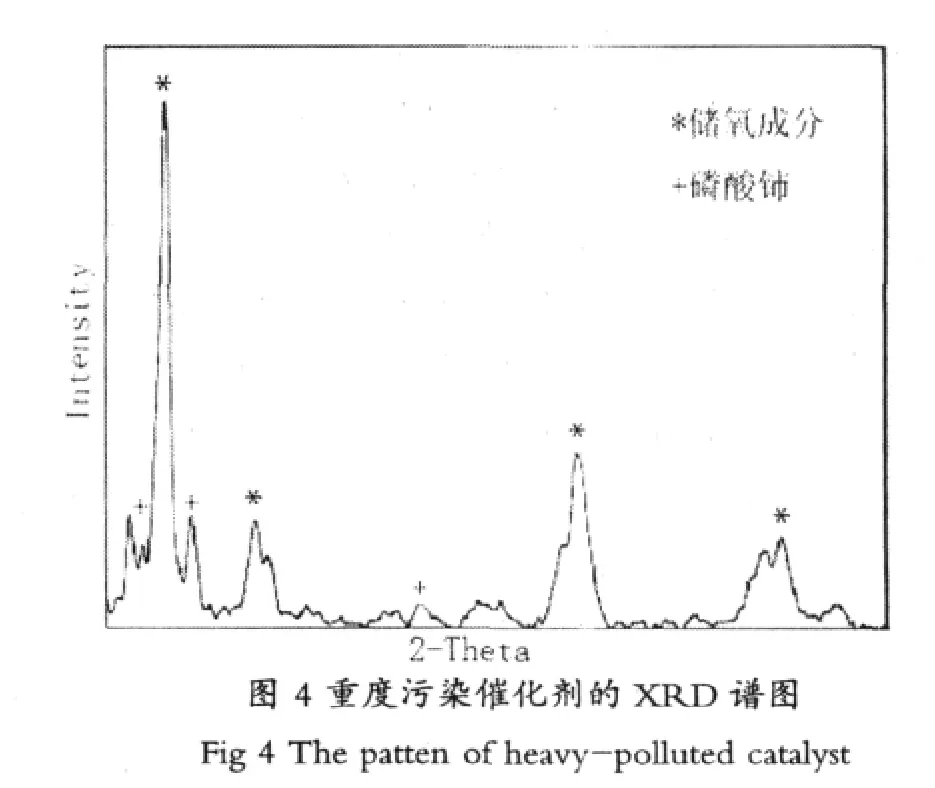
潤滑油在氣缸內燃燒,也可能導致P,Zn和Ca的化合物沉積在三效催化劑的表面,沉積物越多會減少催化劑的活性位,或與催化劑中的活性成分反應。例如,汽車尾氣催化劑配方中的Ce與P反應生成磷酸鈰,XRD分析重度污染的催化劑見圖4,可以明顯看到出現磷酸鈰的衍射峰。近來,汽車尾氣催化劑設計引入了新材料,具有更強的熱穩定和抗化學失活能力。而且強效促進劑和制備工藝的改進已經取得進展,為減少催化劑的中毒,潤滑油的P含量已經降低到不會在催化劑表面沉積。ZDDP的替代品硼基添加劑不影響催化劑的性能。
4 催化劑的失活動力學
催化劑的失活動力學方程主要是溫度,時間,壓力和不同物質濃度的函數。催化劑活性的改變可能是一種或多種過程影響的結果。對于催化劑的失活,動力學來源于在等溫情況下活性金屬的表面積和時間之間的對比,許多學者已經嘗試推導汽車尾氣催化劑的失活動力學。方程(3)是Wanke,Bartholomew等[4]推導的最初的失活動力學的方程。
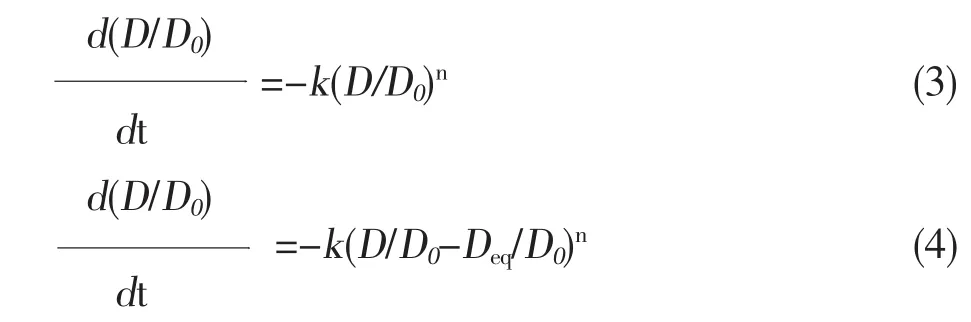
D—貴金屬的分散度(貴金屬的比表面積)
D0—貴金屬最初的分散度(貴金屬原始的比表面積)
k—失活的動力學常數
n—失活反應的反應級數
Deq—貴金屬最終的分散度(采用漸近法得到)
在方程(3)中,k值隨著燒結時間和分散度的改變而變化,近來,Bartholomew等[4]提出了更為精確的表達,通過采用漸近法,引入了Deq(從典型的分散度與時間曲線上計算得到)。方程(4)可以定量的比較,溫度,時間和氣體對催化劑載體失活速率的影響。對研究催化劑的失活提供重要的理論依據。
5 汽車三效催化劑的改性
為了延長催化劑的使用壽命,須改善催化劑的活性組分和涂層材料的熱穩定性,使催化劑具有更強的抗老化能力,目前研究的主要是對催化劑的涂層材料進行改性,通常是加入一些添加劑,包括稀土氧化物、堿土金屬氧化物、二氧化硅及其它氧化物。
5.1 稀土氧化物和γ-Al2O3的作用機制
關于兩者的作用機制很多學者提出了不同的解釋,謝有暢等[8]認為稀土在改性γ-Al2O3時,占據了其活性中心,降低了氧化鋁的表面能,能有效的抑制其燒結;赫崇衡等[9]研究發現,稀土與γ-Al2O3相互作用生成了尖晶型化合物,因此具有很好的熱穩定性能;Church等[12]研究了氧化鋁在老化后的表面積與稀土離子(La3+、Ce4+、Yb3+、Pr3+、Sm3+)半徑之間的關系,其中La3+的半徑最大,改性后的氧化鋁最穩定。Schaper等[16]研究表明La3+在Al2O3表面形成LaAlO3層,阻止了γ-Al2O3由于表面擴散而引起的燒結,從而穩定氧化鋁。Ozawa Masaruni等[10]認為,在高溫下,La3+可以進入到Al2O3的晶格中,占據密堆層中氧離子形成的空隙,從而降低了Al2O3晶格中的離子活性,抑制表面或體相Al3+和O2-的擴散和α-Al2O3的形成。
5.2 堿土金屬氧化物和γ-Al2O3的作用機制
目前能夠穩定氧化鋁的堿土金屬氧化物有BaO、CaO、SrO。其中BaO穩定作用最強,SrO居中,CaO的穩定性能最差。Sepulveda-Escribano等[14]認為BaO的高溫穩定作用被認為是由于BaO的引入改變了Al2O3的結構,形成了形成了鋁酸鹽相(BaAl2O4和BaAl12O19)。該鋁酸鹽相可以有效的阻止A13+體相擴散,且BaO還消除了引起燒結的可移動的物種AlOH,降低了失活的動力學速度,抑制了失結。
5.3 SiO2和γ-Al2O3的作用機制
SiO2對Al2O3的穩定作用要強于稀土和堿土金屬氧化物。研究發現通過溶膠-凝膠和超臨界干燥制備的SiO2改性的氧化鋁,在高溫下老化仍能保持較高的比表面積。Amato[15]認為SiO2能生成玻璃狀表面層而抑制Al2O3相變的發生。Beguin等[16]認為SiO2的穩定作用是由于難移動的Si-OH取代了Al2O3表面易移動的Al-OH。并在脫羥基過程中形成了Si-O-Si或Si-O-Al橋,消除Al2O3表面的陰離子空穴。這與Johnson的模型解釋似乎有點分歧,Johnson認為隨著羥基的不斷脫去,顆粒間形成一個規整的頸狀區域,小顆粒粘結成大顆粒而導致表面積劇減。因此他們認為氧化鋁表面的羥基被氧化硅取代,很難脫去水分子,從而抵制了氧化鋁的燒結。從材料學的角度看,Si和Al的電負性雖然相差很大,但是Si-O鍵和Al-O鍵的鍵長和鍵性相似,因此也能形成固溶體,從而穩定了氧化鋁。雖然作用機理還不是很明確,但是通過氧化硅改性后的氧化鋁具有良好的抗高溫老化性能。
5.4 稀土元素與貴金屬之間的作用機制
稀土元素除了能改善氧化鋁的熱穩定性,還可與貴金屬之間形成協同作用力,這種作用力強于貴金屬和氧化鋁之間的作用力。通過上文知道稀土La可以提高氧化鋁的熱穩定性能,事實還能改善Pd催化劑性能,能夠提高Pd催化劑對NOx還原活性和選擇性。同時可以促進HC對CO還原NO反應的反生。貴金屬Rh與鈰鋯因溶體之間協同作用非常明顯,在反應過程中Rh可以從固溶體的表相遷移至其體相中來抑制固溶體的燒結[17],由于Rh的離子半徑相對于Pt和Pd而言比較小。此外,貴金屬和鈰鋯固溶體之間的相互作用還能促進NO和CO反應[18]。NO的解離吸附主要發生在固溶體的氧空位上,生成的表面物種溢流到貴金屬上,加速了CO的氧化,同時還使得固溶體的氧空位再生。
以上論述了如何提高涂層材料氧化鋁和貴金屬的抗老化性能,主要涉及到一些稀土和堿土材料,雖然在實踐中有了一些非常重要的應用,但是還存在著許多的不足,許多復雜的作用機理還未弄清楚。
6 結語
汽車三效催化劑的失活是當前研究的熱點,也是一迫切需要解決的問題。由于貴金屬儲量的日益匱乏,如何減少貴金屬的用量,延長催化劑的使用壽命是當前尾氣催化劑領域所面臨的另一挑戰,隨著燃油中有毒物質的日益減少,催化劑的失活最大問題是熱失活,研究和開發新型的耐高溫老化材料是目前最主要的問題,從而能更好的提高催化劑的使用壽命。
[1]Beck D D,Sommers J W,Dimaggio C L.Impact of sulfur on model palladiun-only catalysts under simulated threeway operation[J].Appl.Catal,1994,3:205-227.
[2]Chen G,Chou W T.The sorption of hydrogen on palladium in a flow system[J].Appl Catal,1983,8(1):389.
[3]Bartholomew C H.Mechanisms of catalyst deactivation[J].Appl Catal A:General,2001,212:17-60.
[4]Bartholomew C H,Fuentes G A.Sintering and redispersion of supported metals:Perspectives from the literature of the past decade[J].Studies in Surface Science and Catalysis,1997,111:585-592.
[5]Burtin P,Brunelle J P.Influence of surface area and additives on the thermal stability of transition alumina catalyst supports[J].Applied Catalysis,1987,34:225-238.
[6]劉勇,陳曉銀.氧化鋁熱穩定性的研究進展[J].化學通報,2001,2:65-69.
[7]Johnson M F.J.Catal.,1990,123:245.
[8]謝有暢,錢民協,唐有祺.添加La2O3對甲烷催化劑中鎳分散度和熱穩定的影響[J].中國科學B輯,1983,9:788-795.
[9]赫崇衡,張文敏,汪仁,等.稀土修飾Al2O3的表面積熱穩定性[J].物理化學學報,1996,12(11):971-975.
[10]Ozawa M,Kimurta M,Akioisogai.Room temperature oxidation of LaNi5 and the reaction mechanisms of initial hydrogen absorption by LaNi5[J].J.Less-Common Metals,1990,62:297-305.
[11]Lassi,Ulla.Deactivation correlations of Pd/Rh threeway catalyst designed EuroⅣEmission Limits.Effect of ageing atmosphere,temperature and time[M].Acta Univ.Oul.C 180,2003.
[12]Church J S,Cant N W,Trimm D L.ChemInform Abstract:Surface Area Stability and Characterization of a Novel Sulfate-Based Alumina Modified by Rare Earth and Alkaline Earth Ions[J].Appl.Catal.A,1994,107:269.
[13]Rokosz M J,Chen A E,Lowe-Ma C K,et al.Characterization of phosphorus-poisoned automotive exhaust catalysts[J].Appl.Catal.B Environ.2001,33:205.
[14]Sepulveda-Escribano A,Primet M,Praliand H.Influence of the preparation procedure and of the barium content on the physicochemical and catalytic properties of barium-modified platinum/alumina catalysts[J].Appl.Catal.A,1994,108:221.
[15].Navarro R M,álvarez-Galván M C,Sánchez-Sánchez M C,et al.Production of hydrogen by oxidative reforming of ethanol over Pt catalysts supported on Al2O3 modified with Ce and La[J].Appl.Catal.B,2005,55:229-241.
[16]Beguin B,Garbowski E,Drimet M.Pd/SiC Catalysts:Characterization and Catalytic Activity for the Methane Total Oxidation[J].J.Cata.,1991,127:595.
[17]Fornasiero P,Kapar J,Graziani M.Redox behavior of high surface area Rh-loaded Ce0.5Zr0.5O2mixed oxide[J].J.Catal.,1997,176:576-584.
[18]Meriani S.Metastable Tetragonal CeO-ZrO solid solution[J].J.Phys.Suppl.,1986,47:485-490.
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
