基于抗氧化活性的龍眼核標準湯劑提取工藝研究
2025-02-15 00:00:00蔣佳麗王藝潔謝譚芳王志萍
中國藥房 2025年3期
關鍵詞龍眼核;標準湯劑;抗氧化活性;正交實驗;提取工藝
龍眼核為無患子科Sapindaceae植物龍眼DimocarpuslonganLour.的干燥成熟種子,性平,味微苦、澀,具有止血定痛、理氣化濕的功效,可用于創傷出血、癭疾、瘰疬、疝氣等疾病的治療[1]。由于龍眼核未被《中國藥典》收錄,且暫無質量標準,造成其質量控制難度增加、藥效難以保證、臨床應用受限以及廣大患者權益難以保障等問題。據統計,全國每年作為加工副產品而被丟棄的龍眼核超過2萬噸[2],給環境造成了巨大壓力。研究表明,龍眼核中含有豐富的多酚類、黃酮類等抗氧化活性成分[3],這些抗氧化活性成分不僅可通過活化凝血因子激活凝血系統從而改善出血癥狀[4],還可通過清除體內自由基,減少細胞氧化損傷,起到減輕炎癥反應和緩解炎癥反應引起的疼痛的作用[5]。中醫理論認為,氣機不暢、濕邪阻滯會引起自由基代謝紊亂,導致機體過氧化和抗氧化失衡,具體表現為氧自由基增加、超氧化物歧化酶(superoxidedismutase,SOD)減少、丙二醛(malondialdehyde,MDA)增加[6]。龍眼核中的抗氧化成分有較強的自由基清除能力,有助于保持機體氣機的通暢和促進濕氣的運化,這也提示龍眼核藥用價值較大。
標準湯劑是一種以中醫理論為基礎,結合現代制藥技術而形成的標準化提取制劑,具有無輔料干擾、質量穩定、療效可控等優點,可以保障用藥的準確性和劑量的一致性[7],是配方顆粒以及其他劑型的基礎。在中藥提取工藝研究中,加水量、提取時間、提取次數對中藥有效成分的溶出均具有一定的影響[8]。為全面考察龍眼核標準湯劑工藝合理性,本研究采用單因素實驗結合正交實驗,以出膏率、沒食子酸含量、柯里拉京含量、鞣花酸含量及自由基清除能力為評價指標,結合層次分析法(analytichierarchyprocess,AHP)法和基于指標相關性的權重賦權系數(criteriaimportancethoughinter-criteriacorrelation,CRITIC)法進行提取工藝研究,旨在為龍眼核標準湯劑的質量控制和臨床應用奠定基礎。
1 材料
1.1 主要儀器
本研究所用主要儀器包括LC-2030Plus型高效液相色譜(HPLC)儀(日本Shimadzu公司)、SQP型萬分之一電子天平(北京賽多利斯科學儀器有限公司)、XSR205DU/A型十萬分之一電子天平(瑞士Mettler-Toledo公司)、KQ-800DE型數控超聲波清洗器(昆山市超聲儀器有限公司)、HWS-28型電熱恒溫水浴鍋(上海齊欣科學儀器有限公司)、InfiniteM200pro型多功能酶標儀(瑞士Tecan公司)、UPH-Ⅳ-20TN型優普系列超純水機(四川優譜超純科技有限公司)、SCIENTZ-12N型冷凍干燥機(寧波新芝生物科技股份有限公司)等。
1.2 主要藥品與試劑
本研究所用龍眼核藥材于2023年8月采集于福建莆田,經廣西中醫藥大學藥學院田慧教授鑒定為無患子科Sapindaceae植物龍眼D.longanLour.的干燥成熟種子。
對照品沒食子酸(批號MUST-22112411,純度99.96%)、柯里拉京(批號MUST-23060211,純度99.99%)、鞣花酸(批號MUST-23033114,純度99.89%)均購自成都曼斯特生物科技有限公司;2,2′-聯氮雙(3-乙基苯并噻唑啉-6-磺酸)二胺鹽[2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonicacid)ammoniumsalt,ABTS]試劑(批號B2308190,純度98%)購自上海阿拉丁生化科技有限公司;1,1-二苯基-2-苦基肼(1,1-diphenyl-2-picryl-hydrazylradical,DPPH)試劑(批號S041S224966,純度98%)購自上海源葉生物科技有限公司;乙腈、甲醇、磷酸均為色譜純,水為純化水。
2 方法與結果
2.1 出膏率的測定
將龍眼核標準湯劑濃縮至500mL,精密吸取濃縮液10mL到已干燥至恒重的蒸發皿中,蒸干,再于105℃下干燥3h,轉移至干燥器中冷卻30min,迅速稱質量,按下片質量,m1為空皿恒重,m2為含濃縮液蒸發皿恒重后質量)。
2.2 指標成分含量測定
2.2.1 色譜條件
采用島津ShimNexCSC18(250mm×4.6mm,5μm)色譜柱,以乙腈(A)-0.1%磷酸溶液(B)為流動相進行梯度洗脫(0~10min,5%A;10~20min,5%A→13%A;20~50min,13%A→17%A;50~60min,17%A→20%A;60~80min,20%A→50%A;80~90min,50%A→5%A);流速為1.0mL/min;柱溫為30℃;檢測波長為270nm;進樣體積為10μL。
2.2.2 混合對照品溶液的制備
精密稱取沒食子酸、柯里拉京、鞣花酸對照品適量,加甲醇溶解,制成質量濃度依次為413.43、409.56、531.81μg/mL的混合對照品溶液。
2.2.3 凍干粉供試品溶液的制備
取龍眼核100g,加8倍水浸泡30min后提取30min,收集濾液;藥渣加6倍水提取20min,合并濾液并濃縮至500mL。將濃縮液于-80℃冰箱中預凍12h,真空冷凍干燥機中凍干,得龍眼核標準湯劑凍干粉。取凍干粉0.2g,精密稱定,置于50mL具塞錐形瓶中,加甲醇25mL,精密稱定,超聲15min,放冷,加甲醇補足減失的質量,搖勻,過0.45μm微孔濾膜,取續濾液,即得凍干粉供試品溶液。
2.2.4 方法學考察
參照2020年版《中國藥典》(四部)分析方法驗證指導原則進行方法學考察。系統適用性試驗結果顯示,凍干粉供試品溶液與混合對照品溶液在相同保留時間處有相同色譜峰出現(圖略),各待測成分色譜峰與相鄰色譜峰的分離度均大于1.5,理論板數均不少于5000,且空白溶液(甲醇)對測定無干擾,表明本方法系統適用性較好。以各對照品質量濃度為橫坐標(X)、峰面積為縱坐標(Y)繪制標準曲線,得沒食子酸、柯里拉京、鞣花酸的線性回歸方程分別為Y=32191.00X+36752.00(r=0.9999)、Y=19671.00X+5411.20(r=1.0000)、Y=50255.00X-1512.90(r=0.9997),線性范圍分別為10.33~413.43、10.24~409.56、13.30~531.81μg/mL。上述3種成分精密度試驗的RSD分別為0.81%、0.62%、0.68%(n=6);重復性試驗的RSD分別為1.36%、1.01%、0.90%(n=6);穩定性試驗的RSD分別為1.44%、1.67%、1.73%(n=6);平均加樣回收率分別為97.04%、94.73%、101.04%,RSD分別為2.21%、2.24%、2.09%(n=6)。
2.3 DPPH自由基清除實驗
2.3.1 溶液的配制
精密稱取龍眼核標準湯劑凍干粉適量,加甲醇制成質量濃度為0.05、0.1、0.2、0.4、0.8、1mg/mL的供試品溶液。取DPPH適量,加甲醇制成質量濃度為0.3mg/mL的DPPH甲醇溶液。上述溶液均避光保存,備用。
2.3.2 DPPH自由基清除率的測定
參考文獻[9]方法,取“2.3.1”項下不同質量濃度的供試品溶液與DPPH甲醇溶液各100μL于同一孔中,作為實驗組。以甲醇溶液代替DPPH甲醇溶液,其余操作同實驗組,作為對照組;以甲醇溶液代替供試品溶液,其余操作同實驗組,作為標準組。將各組樣品置于暗處反應30min,使用酶標儀于517nm波長處測定各組吸光度(A),按下式計算DPPH自由基清除率:DPPH自由基清除率=1-(A2-A1)/A(0式中,A2為實驗組A,A1為對照組A,A0為標準組A)。運用GraphPadPrism8.3.0軟件計算樣品對DPPH自由基的半數清除濃度(halfclearanceconcentration,IC50),記為DPPH自由基IC50。
2.4 ABTS自由基清除實驗
2.4.1 溶液的配制
精密稱取龍眼核標準湯劑凍干粉適量,加甲醇制成質量濃度為0.05、0.1、0.2、0.4、0.8、1mg/mL的供試品溶液。將3.84mg/mL的ABTS溶液與1.34mg/mL的過硫酸鉀溶液按體積比1∶1混合,于室溫、暗處靜置16h后得ABTS自由基儲備液。使用前,以磷酸鹽緩沖液(PBS,pH7.4)稀釋約20倍,得ABTS自由基工作液。
2.4.2 ABTS自由基清除率的測定
參考文獻[10]方法并適當修改后進行測定。取“2.4.1”項下不同質量濃度的供試品溶液20μL和ABTS自由基工作液180μL于同一96孔板中,作為實驗組。以PBS代替ABTS工作液,其余操作同實驗組,作為對照組。以PBS代替供試品溶液,其余操作同實驗組,作為標準組。將各組樣品置于暗處反應30min,使用酶標儀于734nm波長處測定各組吸光度(A),按下式計算ABTS自由基清除率:ABTS自由基清除率=1-(A2-A1)/A0(式中,A2為實驗組A,A1為對照組A,A0為標準組A)。運用GraphPadPrism8.3.0軟件計算樣品對ABTS自由基的IC50,記為ABTS自由基IC50。
2.5 單因素實驗篩選提取工藝條件
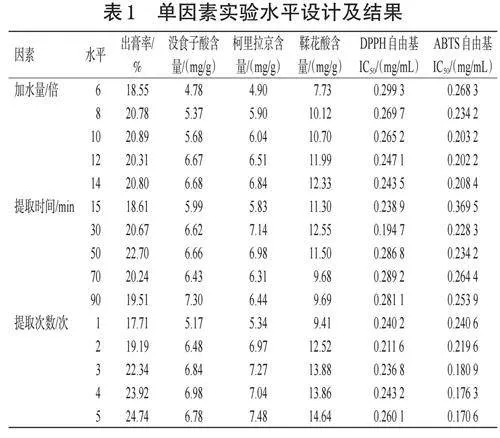
稱取龍眼核藥材,每份100g,搗碎表皮及種仁,浸泡30min,按表1水平分別對加水量、提取時間、提取次數進行系統考察。將龍眼核標準湯劑濃縮至500mL,分別按“2.1”~“2.4”項下方法測定出膏率、指標成分含量、DPPH自由基IC50和ABTS自由基IC50。結果(表1)顯示,在加水量為10~14倍時,出膏率變化不大,沒食子酸含量、柯里拉京含量、鞣花酸含量增加較為明顯,DPPH、ABTS自由基IC50較低,故選擇加水量為10~14倍進行正交實驗。在提取時間為50min時出膏率最高,沒食子酸含量在提取90min時最高,提取時間為30min時柯里拉京含量、鞣花酸含量最高并且DPPH、ABTS自由基IC50最低,結合時間成本,選擇提取時間為15~50min進行正交實驗。出膏率隨提取次數的增加而升高,沒食子酸含量在提取4次時最高;柯里拉京含量、鞣花酸含量均在提取5次時最高,但與提取2~4次含量相差不大;ABTS自由基IC50變化趨勢亦如此,但在提取2次時DPPH自由基IC50最低。綜合時間、耗能等因素選擇提取2~4次進行正交實驗。
2.6 AHP-CRITIC法結合正交實驗優選提取工藝
2.6.1 AHP法計算權重
多項研究結果表明,龍眼核具有良好的抗氧化活性[11―12],為確保龍眼核標準湯劑的抗氧化效果,故將龍眼核標準湯劑對DPPH、ABTS自由基的清除能力作為第一重要指標。多酚類成分為龍眼核抗氧化作用主要化學成分[3],故將龍眼核標準湯劑中3種多酚類成分(沒食子酸、柯里拉京、鞣花酸)含量作為第二重要指標。出膏率雖然是判斷提取工藝穩定性的重要指標,但未直接反映龍眼核標準湯劑抗氧化效果和化學信息[13],故將其作為第三重要指標。按DPPH自由基IC50=ABTS自由基IC50>沒食子酸含量=柯里拉京含量=鞣花酸含量>出膏率的順序,采用SPSSPRO在線系統(https://www.spsspro.com/)對評價指標進行兩兩比較,構建判斷矩陣并進行一致性評價。一致性檢驗結果顯示,最大特征根為6,一致性指標為0,平均隨機一致性指標為1.25,一致性比例=0<0.1,表明該判斷矩陣一致性良好[14]。根據該比較矩陣,得到出膏率、沒食子酸含量、柯里拉京含量、鞣花酸含量、DPPH自由基IC50、ABTS自由基IC50的權重分別為7.692%、15.385%、15.385%、15.385%、23.077%、23.077%。故AHP法的綜合得分=出膏率實測值/最大值×7.692%+沒食子酸含量實測值/最大值×15.385%+柯里拉京含量實測值/最大值×15.385%+鞣花酸含量實測值/最大值×15.385%+DPPH自由基IC50最小值/實測值×23.077%+ABTS自由基IC50最小值/實測值×23.077%。
2.6.2 CRITIC法計算權重
根據文獻[15]方法,先對數據予以標準化處理,然后采用SPSSPRO在線系統(https://www.spsspro.com/)計算各指標權重,得到出膏率、沒食子酸含量、柯里拉京含量、鞣花酸含量、DPPH自由基IC50、ABTS自由基IC50的權重分別為19.823%、12.663%、20.759%、12.040%、14.754%、19.963%。故CRITIC法的綜合得分=出膏率實測值/最大值×19.823%+沒食子酸含量實測值/最大值×12.663%+柯里拉京含量實測值/最大值×20.759%+鞣花酸含量實測值/最大值×12.040%+DPPH自由基IC50最小值/實測值×14.754%+ABTS自由基IC50最小值/實測值×19.963%。
2.6.3 AHP-CRITIC法計算綜合權重
根據AHP法、CRITIC法所得權重計算綜合權重(G綜合)∶G綜合ij=(WAHPij×WCRITICij)/Σ(WAHPij×WCRITICij);式中,WAHPij表示用AHP法計算得到的權重,WCRITICij表示用CRITIC法計算得到的權重,i、j分別表示指標i和指標j。經計算,出膏率、沒食子酸含量、柯里拉京含量、鞣花酸含量、DPPH自由基IC50、ABTS自由基IC50的G綜合分別為9.224%、11.784%、19.320%、11.206%、20.597%、27.869%。
2.6.4 3種權重方法比較
分別按AHP法、CRITIC法及AHP-CRITIC法計算綜合得分,并采用SPSS27.0.1軟件對3種方法計算所得的綜合得分進行相關性分析。經計算,AHP法與CRITIC法、AHP法與AHP-CRITIC法、CRITIC法與AHP-CRITIC法的相關系數分別為0.983、0.999、0.986,三者兩兩比較均具有顯著相關性(P<0.01),說明3種評分方法具有良好的一致性。同法對3種方法的權重進行相關性分析,結果顯示,AHP法與CRITIC法權重的相關系數為-0.119,且不具有顯著性(P=0.823>0.05),表明二者所呈現的信息不存在重疊。由于AHP-CRITIC法同時兼顧主觀判斷與客觀數據,因此本研究最終采用AHP-CRITIC法計算綜合得分,即綜合得分=出膏率實測值/最大值×9.224%+沒食子酸含量實測值/最大值×11.784%+柯里拉京含量實測值/最大值×19.320%+鞣花酸含量實測值/最大值×11.206%+DPPH自由基IC50最小值/實測值×20.597%+ABTS自由基IC50最小值/實測值×27.869%。
2.6.5 正交實驗設計
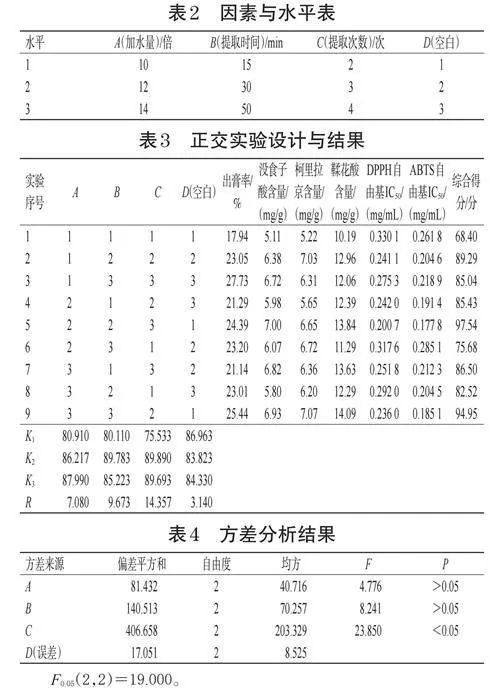
根據前期單因素結果,以加水量(A)、提取時間(B)、提取次數(C)為影響因素,以出膏率、沒食子酸含量、柯里拉京含量、鞣花酸含量、DPPH自由基IC50、ABTS自由基IC50為評價指標,以AHP-CRITIC法計算各評價指標綜合得分,采用L(934)正交實驗優化龍眼核標準湯劑提取工藝。因素與水平見表2,正交實驗設計與結果見表3,方差分析結果見表4。
由表3可知,各因素對提取工藝的影響順序為C>B>A,且A3>A2>A1、B2>B3>B1、C2>C3>C1。由表4可知,因素C對提取工藝的影響顯著(P<0.05),而因素A和B的影響不顯著(P>0.05)。故確定最佳提取工藝為A3B2C2,即第1次提取加14倍水,提取30min,第2、3次提取均加12倍水,提取20min。
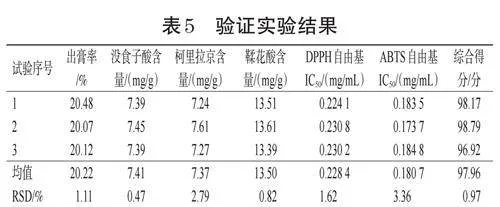
2.6.6 提取工藝驗證
按“2.6.5”項下工藝制備3批樣品,進行工藝驗證,并按“2.1”~“2.4”項下方法測定各項指標,結果見表5。由表5可知,3批驗證實驗數據一致性較高,證明所選的提取工藝穩定可靠,具備良好的可重復性。
3 討論
3.1 色譜條件
本研究前期分別對凍干粉的提取條件(提取方式、提取溶劑種類、提取溶劑用量)以及液相色譜條件進行了系統考察。最終確定凍干粉最佳提取條件為25mL甲醇超聲提取15min。色譜條件考察結果顯示,在270nm波長處各指標成分吸收好,在流動相體系為乙腈-0.1%磷酸溶液時基線平穩、各峰峰形尖銳且對稱,當柱溫為30℃、流速為1.0mL/min時各色譜峰分離度較好。
3.2 龍眼核破碎程度
傳統煎煮認為“逢殼必搗,逢子必破”。龍眼核為龍眼干燥成熟種子,呈類球形,表皮呈紅褐色,剖開后有2片子葉,質堅硬。本研究前期對龍眼核破碎程度進行了考察,在僅搗碎龍眼核表皮、不搗碎子葉的情況下,龍眼核標準湯劑出膏率為6.54%,沒食子酸、柯里拉京、鞣花酸含量分別為5.26、2.89、5.50mg/g,DPPH、ABTS自由基的IC50分別為0.2844、0.3700mg/mL,該情況下多酚類成分含量少,抗氧化效果較差,也不易凍干。結合實驗現象和文獻[16]分析,可能原因是此時提取液中化學成分多為龍眼核多糖,使得提取液較為黏稠,從而導致樣品在凍干和升華過程中擴散困難,影響凍干效率;當龍眼核表皮及子葉均被搗碎時,溶劑更易進入藥材內部,促使多酚類有效成分溶出,增強了抗氧化效果,也更易凍干,此時出膏率為18.44%,沒食子酸、柯里拉京、鞣花酸含量分別為6.42、6.65、11.38mg/g,DPPH、ABTS自由基的IC50分別為0.2466、0.2433mg/mL。故本研究在進行提取之前,將龍眼核表皮及子葉搗碎,并浸泡30min,以便有效成分溶出。
3.3 評價指標及方法選擇原因
以往提取工藝研究多以出膏率、指標成分含量作為評價指標[17―18],但中藥成分復雜,僅通過控制出膏率及有效成分含量無法確保得到的是最佳提取工藝。因此,為了更全面、深入地進行評價,本研究在以關鍵指標含量和出膏率為評價指標的基礎上,將2個抗氧化藥效實驗結果作為主要評價指標。AHP法是一種主觀的評價方法,主要由研究者根據評價指標的重要性進行打分確定權重,CRITIC法則基于數據自身的變異性和指標間的沖突性來確定權重[19]。本研究將AHP法與CRITIC法相結合,相較于單一的評分方法,既體現了研究者的主觀判斷,也反映了數據的客觀特征,可以減少單一評分方法所帶來的偏差和局限性,使評價結果更為科學、合理。再通過對加水量、提取時間、提取次數3個因素的綜合考察,優化出龍眼核標準湯劑最佳提取工藝為第1次提取加14倍水,提取30min;第2、3次提取均加12倍水,提取20min。用此條件進行3批驗證實驗,平均綜合得分為97.96分,RSD為0.97%,表明該工藝穩定、可行。
綜上,本研究基于抗氧化活性,采用AHP-CRITIC法結合正交實驗優化的龍眼核標準湯劑提取工藝穩定可行。本研究結果可為龍眼核標準湯劑的深入開發利用提供依據。
