高性能鋰硫電池充放電過程的調控與機制分析
2024-12-16 00:00:00孫琪何文輝逯樂慧
分析化學 2024年11期
關鍵詞:順鉑

摘要 鋰硫電池的充放電循環為16 電子轉移過程,使得硫正極材料的比容量高達1675 mAh/g。然而,緩慢且復雜的動力學過程也導致了可溶性多硫化物的穿梭效應和低活性物質利用率。鑒于此,本研究采用順鉑作為氧化還原介質,通過多硫化物與順鉑間自發的氧化還原反應,調控鋰硫電池充放電過程,并同時以順鉑為分子探針識別鋰硫電池活性物質氧化還原反應路徑。研究結果表明,引入順鉑可加快電荷轉移動力學過程,調控氧化還原路徑,并實現Li2S2 到Li2S 的深度轉化。引入順鉑后的鋰硫電池的最大比容量達到1290 mAh/g,循環1000 圈后,平均每圈容量衰減僅為0.017%,并且在具有實際應用價值的低電解液/硫比(2.5 μL/mg)的軟包電池中實現了318.8 Wh/kg 的高能量密度。本研究通過原位拉曼光譜、電化學石英晶體微天平技術和質譜等表征手段對順鉑作用機制進行了探究,結果表明,順鉑可通過產生具有氧化還原活性的順鉑/多硫化物復合物調控鋰硫電池反應路徑,使緩慢遲滯的充放電過程被動力學更迅速的Pt4+/Pt2+對的氧化還原過程取代。本研究結果為合理利用功能化氧化還原介體解決鋰硫電池中的關鍵問題提供了新思路。
關鍵詞 鋰硫電池;順鉑;充放電過程;穿梭效應;原位表征技術
基于硫(S)的高天然豐度、高理論鋰化容量(1675 mAh/g)和環境友好等優勢,鋰硫電池已成為新一代極具商業潛力的可充電鋰電池之一[1-2]。然而,鋰硫電池在充放電過程中須歷經多種復雜反應路徑并產生一系列中間產物,這使得完成S8 與Li2S 之間16 電子的氧化還原轉化反應極具挑戰性[3-4]。充放電過程中多硫化物的積累及其遷移過程導致了活性物質損失與電池容量快速衰減,即多硫化物的穿梭效應;此外,多硫化物到Li2S2/Li2S 之間的電化學轉化過程受限于Li2S2/Li2S 固體的絕緣性,這嚴重制約了鋰硫電池理論性能的釋放[5-6]。因此,通過調控鋰硫電池充放電過程抑制穿梭效應,并促進多硫化物到Li2S的轉化反應,對于提高鋰硫電池性能及推動其商業化進程至關重要[7]。
近年來,科研人員一直致力于通過鋰硫電池正極材料的改性與結構設計抑制穿梭效應的研究[8-10]。然而,隨著充放電循環次數累加,此類正極材料表面在絕緣性Li2S2/Li2S 固體的沉積作用下將不可避免地被鈍化[11-12],因此, Li2S2/Li2S 包覆的正極材料表面將傾向以更緩慢的動力學過程向自催化多硫化物轉化,這將進一步加劇多硫化物的穿梭效應[13-14]。
向鋰硫電池電解液中引入電化學活性氧化還原介質是一種可有效抑制鋰硫電池穿梭效應或促進Li2S2/Li2S 氧化還原轉化反應的可行性策略[14]。此氧化還原介質可分為兩類:一類通過生成多種硫醇自由基促進S—S 鍵的斷裂過程[15];另一類對絕緣的Li2S2/Li2S 固體進行化學氧化[16-17]。然而,這兩種介質都被認為是一種單功能添加劑,無法在克服穿梭效應的同時解決Li2S 低轉化率的問題[18]。此外,向鋰硫電池中引入分子探針,對于探究氧化還原機制具有重要意義,例如,將1,3,5-苯三硫酚(BTT)引入鋰硫電池,可在改善循環性能的同時協助探究鋰金屬沉積及硫氧化還原電化學反應機制[19]。因此,尋找合適的氧化還原介質以實現鋰硫電池充放電過程的優化調控與機制探索具有重要意義[20]。
本研究以順鉑為氧化還原介質,通過剪切多硫化物的S—S 鍵促進多硫化物到Li2S 的轉化,同時以其Pt2+活性位點作為分子探針,通過追蹤多硫化物的鏈長變化識別改良充放電反應路徑。在順鉑作用下,鋰硫電池的放電反應路徑S8 2– → S62– → S3·– → Li2S2/Li2S 遠快于S8 2– → S4 2– → Li2S2,而反向的充電反應遵循Li2S2/Li2S → S42– → S62– → S8 路徑。本研究結果表明,順鉑可通過Pt4+/Pt2+快速氧化循環過程替代原有鋰硫體系遲滯的充放電過程,大幅提高扣式鋰硫電池的循環穩定性及庫侖效率。此外,順鉑在鋰硫軟包電池中具有318.8 Wh/kg 的高能量密度,因而具有良好的應用前景。
1 實驗部分
1.1 儀器與試劑
EVOLUTION One Plus 紫外-可見光譜儀和MSQ 單四極桿質譜儀(美國賽默飛世爾科技公司);布勞恩手套箱(德國);WITec alpha 300R 共聚焦拉曼光譜儀(德國);Land CT2001A system 電池測試儀(武漢藍和電子有限公司);P3000A 電化學工作站(美國普林斯頓公司);CHI 440C 電化學工作站(上海辰華公司);旋轉圓盤電極(美國PINE 公司);101A-1E 電熱鼓風干燥箱(上海精宏有限公司);VGESCALABMKLL 電子能譜儀(英國VG Scientific 公司)。
高純硫(S, 99.99%(m/m,下同))、硫化鋰(Li2S, 99.98%)、鋰箔(Li, 99.9%)、雙三氟甲基磺酸亞酰胺鋰(LiTFSI, 99.95%)、乙二醇二甲醚(DME,無水級, 99.5%)、1,3-二氧戊環(DOL,無水級, 99.8%)、聚偏氟乙烯(PVDF)、N-甲基吡咯烷酮(NMP, 99.5%)、硝酸鋰(LiNO3, 99.99%)和順鉑(cis-Pt, 99.5%)等藥品購自西格瑪奧德里奇(上海)貿易有限公司;多壁碳納米管(MWCNTs,≥95%)、二硫化碳(CS2,gt;99%)和炭黑購自上海阿拉丁試劑公司。以上試劑未經任何處理,直接使用。如無特殊說明,實驗用水均為高純去離子水。
1.2 實驗方法
1.2.1 S-MWCNTs 正極復合材料的合成
采用熔融-擴散法合成了負載硫的多壁碳納米管(S-MWCNTs)正極材料。將MWCNTs 與S 粉體按質量比3∶7 混合后,充分研磨30 min,將其溶于CS2 中,充分攪拌,將所得混合物在烘箱中以155 ℃加熱12 h,即得到S-MWCNTs 復合材料。將此S-MWCNTs 復合材料、導電炭黑和PVDF 粘結劑按質量比8∶1∶1 混合于NMP 中,攪拌得到平滑均勻的漿料。將漿料攪拌2 h 后,涂布于鋁箔上,在真空烘箱中以60 ℃干燥12 h,即得到涂覆正極材料的鋁箔片。將鋁箔片裁剪成直徑為12 mm 的圓形電極片,平均每個電極片的硫負載量為1.0 mg/cm2,由此即得扣式電池組裝所需S-MWCNTs 正極復合材料。
1.2.2 Li2Sx 溶液的合成
將Li2S 和S 粉末以對應摩爾比和常規鋰硫電池電解液混合(1 mol/L LiTFSI 的DOL/DME(1∶1, V/V)溶液),制得Li2Sx 溶液。例如,將Li2S 與S 粉末以摩爾比1∶7 加入到LiTFSI/DOL/DME 電解液中,在氬氣氛圍下于60 ℃加熱并充分攪拌,直到混合粉末在其中完全溶解,即可得到Li2S8 溶液。
1.2.3 鋰硫電池的組裝與電化學表征測試
實驗所用扣式電池型號為CR2032,電池的隔膜、負極與電解液分別為Celgard 2400 膜、鋰片和20 μL 含1 mol/L LiTFSI 的DOL/DME(1∶1, V/V)溶液,在充滿氬氣的手套箱中完成電池組裝。所裝配的扣式電池的恒電流充放電測試由Land CT2001A System 進行,測試條件如下:電壓測量范圍為1.7~2.8 V(vs. Li/Li+),在不同倍率(即“C”值, 1 C=1675 mA/g)條件下測量并計算電池的比容量。循環伏安(CV)測試與電化學阻抗譜(EIS)測試均采用普林斯頓P3000A 電化學工作站完成, CV 測試實驗條件如下:電池由開路電壓(OCV)以0.05 mV/s 的速度慢掃至1.0 V,而后回掃至3.0 V。
軟包電池能量密度由式(1)計算得到:
Eg = (Careal × V ) / (m areal)i (1)
其中, Careal、V 和mareal 分別代表軟包電池的面容量、平均輸出電壓(2.1 V)和面平均質量。
1.2.4 原位拉曼光譜測試
原位拉曼光譜實驗基于鋰硫電池的軟包電池進行。本研究使用特制的軟包電池進行拉曼光譜測試,軟包電池上設計有石英窗口,用于收集拉曼信號,所用拉曼激光光源為532 nm。分別以涂覆S 的Al 箔、Li 箔以及有/無順鉑的LiTFSI/DOL/DME 為軟包電池的正極、負極和電解液。將軟包電池以0.05 C 的電流從開路電壓放電至1.6 V,在每個對應待測電壓下保持60 s,以采集此電壓下的拉曼光譜。
1.2.5 原位電化學石英晶體微天平(EQCM)實驗
以CHI 440C 電化學工作站進行原位EQCM 實驗。在測試過程中,將S 電極與Li 金屬電極浸潤在50 μL 1.0 mol/L LiTFSI 的DOL/DME(1∶1, V/V)電解液中,測量在0.1 mol/L 順鉑作用下石英晶體片上涂覆電極的質量變化情況。CV 測試的電壓窗口為開路電壓(OCV)?1.0 V,掃描速度為0.1 mV/s。
EQCM 實驗所得頻率變化數據通過式(2)轉換為電極質量變化[21-22]:
其中, Δf、A、μQ、Q、f0 2 和Δm 分別代表頻率變化、壓電活性晶體面積、石英剪切模量、石英密度、石英晶體的共振頻率和電極質量變化。
2 結果與討論
2.1 順鉑調控鋰硫電池充放電過程設計理念
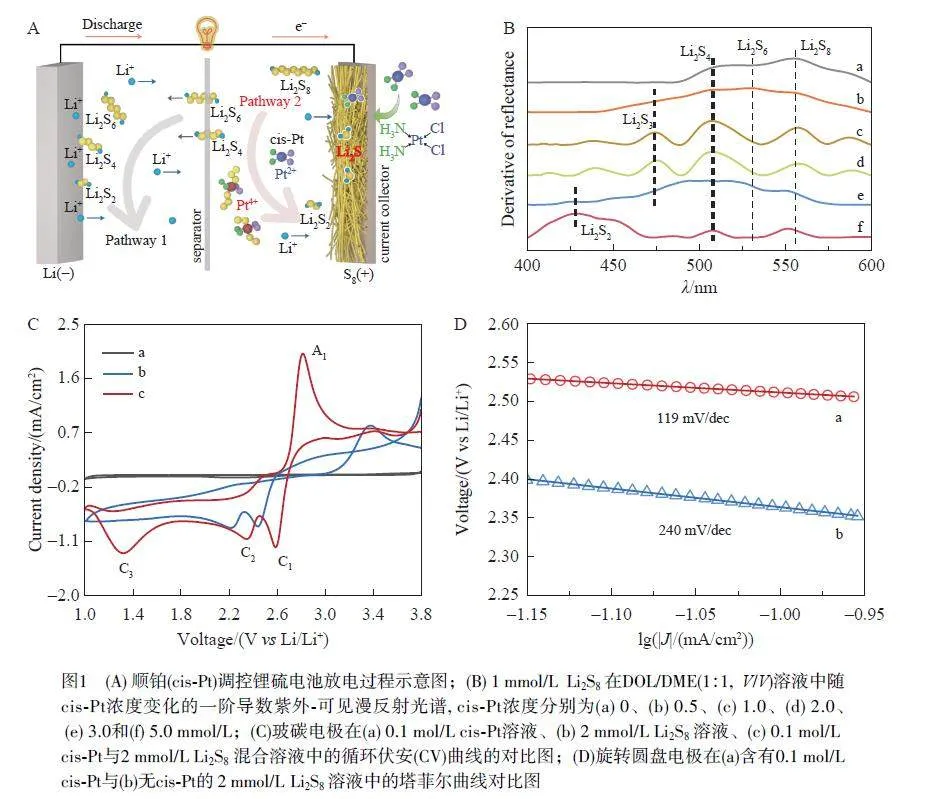
鋰硫電池在放電過程中,活性物質S8 通過雙電子還原過程逐步裂解S—S 鍵,此過程使得S8 在最終轉化為固體Li2S2 和Li2S 產物之前發生了一系列反應,即在最初產生Li2S8,隨后產生表達式為Li2Sn(n=3, 4, 6)的一系列多硫化物。根據文獻[23]報道,可溶性S82– 和Sn2– 的形成相對容易,而Sn2– 轉化為不溶性Li2S2/Li2S 的進程特別緩慢。因此,在放電過程中,可溶性多硫化物不斷地在電解液正極側積累,并擴散至整個電解液區域,產生穿梭效應(圖1A 中途徑1)。為了抑制穿梭效應,本研究在電解液中使用順鉑作為活性添加劑,調控鋰硫電池中的充放電途徑(圖1A 中途徑2)。具有平面四配位的Pt2+中心順鉑可提供2 個電子,分裂多硫化物中的S—S 鍵,并可被氧化成具有2 個軸向Sn2–(n=2, 3)配體的六配位Pt4+中心。隨后, Pt4+被還原為Pt2+并生成Li2S2/Li2S。在這種情況下,可采用反應速度更快的Pt4+/Pt2+氧化還原循環過程取代動力學緩慢的放電過程,從而減輕多硫化物的穿梭效應。
為了驗證上述構想,本研究通過紫外-可見漫反射光譜(UV-DRS)分析了Li2S8 的溶液成分隨加入順鉑濃度(0~5 mmol/L)變化而產生的改變。如圖1B 所示, UV-DRS 一階求導后的數據表明,由于不可避免的化學歧化和中和反應,最初的Li2S8 是Li2S8、Li2S6 和Li2S4 的混合物。添加0.5 mmol/L 順鉑后,在470 nm 處出現了1 個小的肩峰,此峰歸屬為Li2S3[24]。當順鉑濃度增加到1~2 mmol/L 時,在470、505 和560 nm 處出現了3 個分裂峰,分別對應于Li2S3、Li2S4 和Li2S8,而Li2S6 相關峰的消失表明Li2S6 的S—S鍵斷裂速度更快,這與Li2S3 的峰值信號增強所表達的信息一致。由以上分析可知,順鉑促進了LiPSs 向Li2S2 的轉化,并且Li2S6-Li2S3-Li2S2 的轉化比Li2S8-Li2S4-Li2S2 的轉化更迅速。
進一步研究了順鉑在鋰硫體系中改善多硫化物轉化和加速電化學反應的作用。通過CV曲線(圖1C)可以觀察到,在順鉑作用下, S8 溶液在1.37 V 處出現了1 個額外的還原峰C3,對應于S2 2?到S2?的轉化反應,并且A1 峰的存在證明了此反應具有良好的可逆性,順鉑可促進Li2S2-Li2S 的深度轉化以及充電過程。由順鉑作用下S8 溶液的Tafel 圖(圖1D)可見,引入順鉑可使鋰硫體系的Tafel 斜率降低(η=119 mV/dec),表明順鉑可有效加速S8 還原的反應動力學過程。綜上,引入順鉑能夠有效促進鋰硫電池體系遵循更高效的路徑進行放電。
2.2 順鉑調控鋰硫電池性能
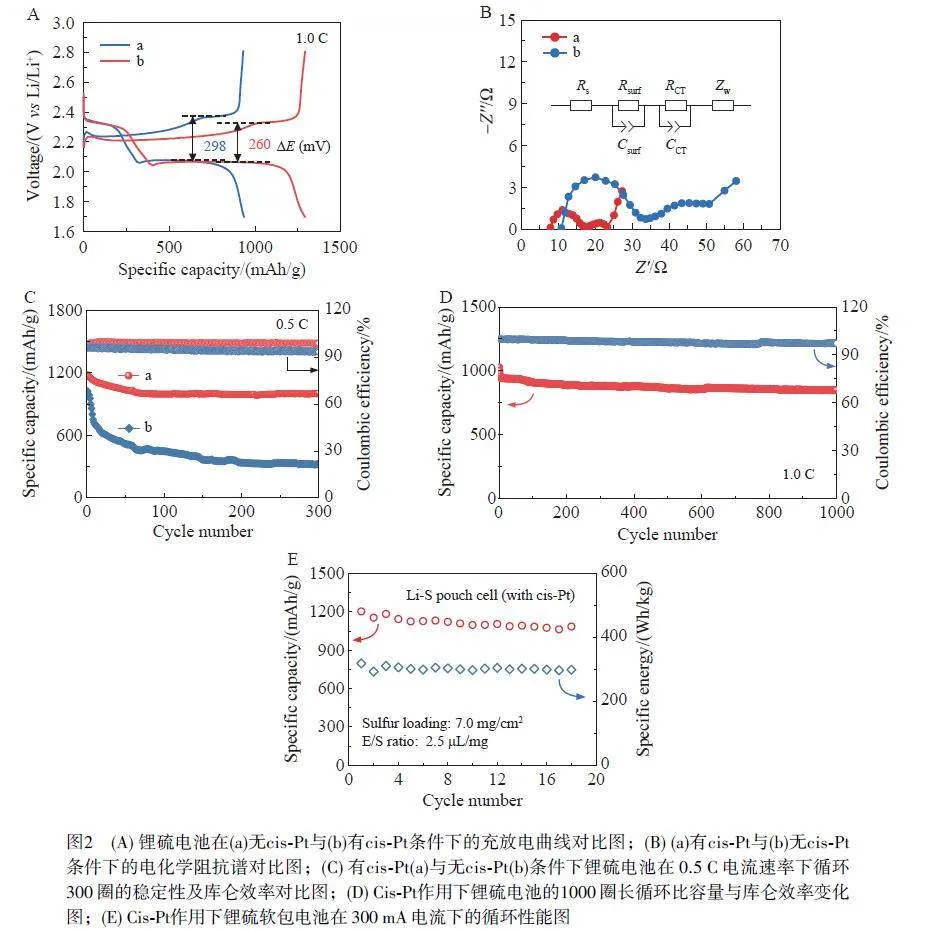
為探究順鉑對鋰硫電池性能的影響,進一步評估了順鉑存在下鋰硫電池的電化學性能。如圖2A 所示,鋰硫電池的恒流充放電曲線在2.4 和2.1 V 處呈現兩個特征放電平臺,分別對應Li2S8-Li2S4 和Li2S4-Li2S2/Li2S 的轉換過程。在1.0 C 下,含順鉑鋰硫電池的比容量為1198 mAh/g,硫的利用率為71.7%,遠高于無順鉑作用下鋰硫電池硫的利用率(58.7%)。此外,順鉑存在時,極化電壓間隙(ΔE)較低(260 mV),顯著低于無順鉑體系(298 mV),表明引入順鉑可使電池體系具有較低的電壓遲滯效應,從而賦予電池良好的循環穩定性。
根據圖2B 所示的電化學阻抗譜(EIS)對比分析,加入順鉑,電池的電荷轉移電阻(Rct)由19.2 Ω顯著降低至8.6 Ω,表明順鉑在提高LiPSs 轉化的電荷轉移動力學方面具有非常獨特的優勢。在0.5 C 電流速率下,含順鉑的鋰硫電池在前50 次循環中的容量衰減輕微,在300 次長循環后仍保持1006 mAh/g 的高比容量,平均每次循環容量衰減0.05%(圖2C);然而,對于無順鉑體系,其比容量則急劇下降至322 mAh/g,在300 次循環中每次循環的容量衰減高達0.23%。此外,含順鉑鋰硫扣式電池循環300 次后庫侖效率仍保持~99%,明顯高于無順鉑鋰硫扣式電池(~93%)。如圖2D 所示,順鉑可使鋰硫電池在長循環1000 圈后仍保持近95%的初始容量,表明其可以有效提高電池循環穩定性。由順鉑在軟包電池中的測試結果(圖2E)可見,初始放電比容量為1203 mAh/g,能量密度為318 Wh/kg,在18 圈內保持了較高循環容量,因而具有良好的應用前景。
2.3 順鉑調控充放電過程的原位表征
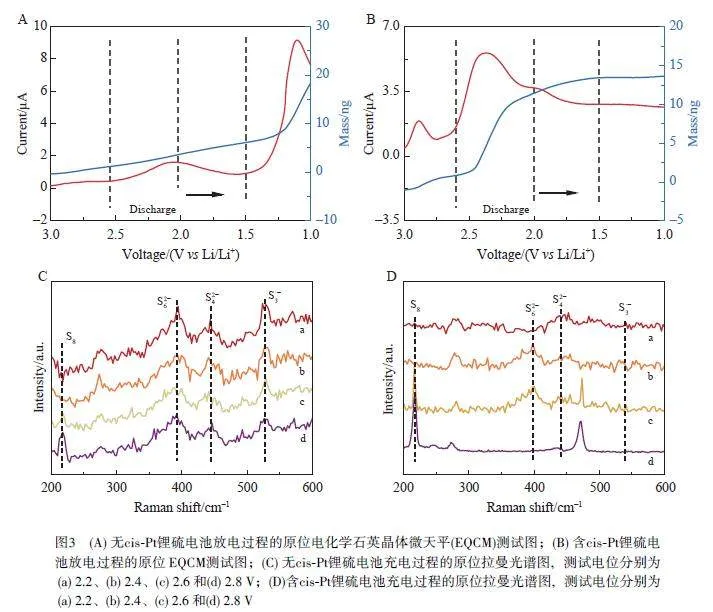
為了驗證順鉑對于鋰硫電池充放電過程的調控作用,進行了原位EQCM 和Raman 光譜表征。如圖3A 所示,在放電初始階段(3.0~2.7 V),含順鉑電池表現出明顯的放電電流,隨后出現質量增加峰,這表明S8 到Li2S8 的鋰化動力學過程迅速。當放電到達2.6~2.1 V 階段時,放電電流急劇增大,鋰硫電池電極質量迅速增加,這表明電極在進行鋰化反應。相比之下,在無順鉑體系的前兩個階段(圖3B),硫電極只顯示出輕微且緩慢的質量增加,表明其緩慢而遲滯的放電反應過程。因此,順鉑可有效促進鋰硫電池的鋰化-放電過程。
由鋰硫電池的原位Raman 表征結果(圖3C 和3D)可見,在充電過程中,無順鉑體系的Li2S2/Li2S 很難被氧化為可溶性多硫化物并最終生成S8;即使電池充電到2.8 V,與多硫化物相關的3 個特征峰仍占主導地位,在220 cm–1 處只出現1 個微弱的S8 信號。與之相反,引入順鉑有利于Li2S2/Li2S 在2.2 V 時轉化為S4 2–,在2.4 V 時轉化為S4 2– 和S6 2–, 2.6 V 時轉化為S6 2– 和S8,并且在2.8 V 時完全轉化為S8。S4 2–、S6 2– 和S8隨充電電位的分階段增長趨勢揭示了鋰硫電池的氧化放電途徑為Li2S2/Li2S → S4 2– → S6 2– → S8。以上原位EQCM 與Raman 光譜表征結果表明,順鉑可高效促進可逆的鋰硫電池充放電過程。
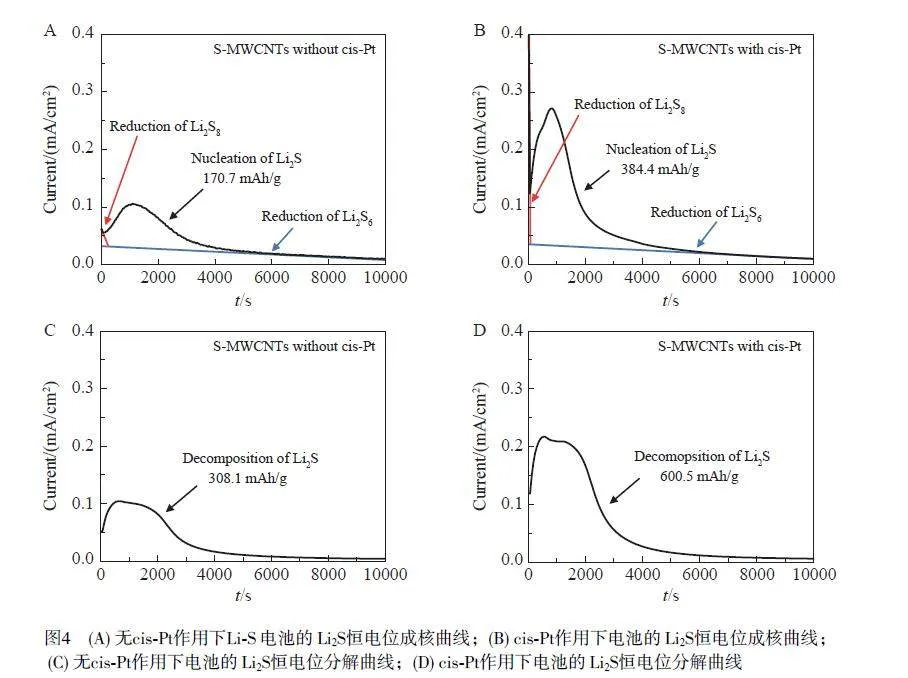
驗證了順鉑對放電產物Li2S 沉積與分解過程的具體作用,如圖4A 和4B 所示,在順鉑作用下,電池表現出更高的Li2S 沉積電流,并且其成核時間相較于對照組更提前。含順鉑體系的Li2S 沉積過程的容量(384.4 mAh/g)是無順鉑體系(170.7 mAh/g)的2 倍以上,表明順鉑可有效促進Li2S 成核,從而加速放電階段S8 的斷鏈過程。此外,如圖4C 和4D 所示,在含順鉑體系中Li2S 分解過程的容量(600.5 mAh/g)相較于無順鉑體系(308.1 mAh/g)顯著增加,表明順鉑可有效促進Li2S 分解。以上結果表明,順鉑可同時促進鋰硫電池高效可逆的循環過程。
2.4 順鉑催化多硫化物轉化機制
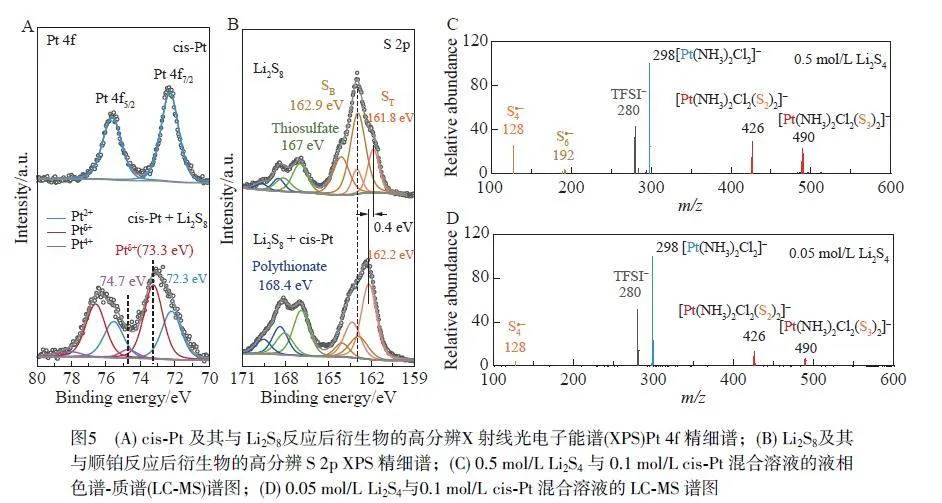
X 射線光電子能譜(XPS)測試結果(圖5A)表明,順鉑與Li2S8 溶液作用后,其Pt 精細譜表現出3 種價態,表明順鉑中Pt2+被S82?氧化成Pt4+,具有2 個額外軸向配位,形成Pt—S 鍵。通過對XPS 高分辨S 2p 精細譜(圖5B)分析可知, SB/ST 比值低于S32?的理論值(1∶2),表明順鉑參與了多硫化物的S—S 鍵斷裂過程,導致生成了S32?和S22?的混合物。液相色譜-質譜(LC-MS)結果(圖5C 和5D)顯示,在順鉑作用下的Li2S4 溶液中,順鉑與多硫化物反應形成了具有2 個Sn 配體的順鉑-多硫化物配合物。以上結果表明,在順鉑引導體系中,以反應速度更快的Pt2+/Pt4+離子對氧化還原循環過程取代了動力學過程緩慢的多硫化物轉化過程,驗證了順鉑催化多硫化物的轉化機制。
3 結論
順鉑可作為一種活性氧化還原介質調控鋰硫電池的充放電過程,在此過程中,緩慢的多硫化物轉化過程被順鉑/多硫化物復合物更快的氧化還原過程所取代,從而有效抑制了穿梭效應。在鋰硫電池中引入順鉑后,其倍率和循環性能顯著提高;此外,軟包電池測試結果表明,順鉑在高能量密度(318.8 Wh/kg)的鋰硫電池中具有應用潛力。原位Raman 光譜和原位EQCM 結果表明,順鉑在調控充放電反應路徑以及促進鋰化-放電動力學方面發揮了重要作用。XPS 和LC-MS 研究結果表明,順鉑和多硫化物之間的氧化還原反應形成了順鉑/多硫化物配合物。本研究闡明了順鉑調控鋰硫電池充放電反應過程的機制,為未來功能化介體的設計提供了參考,對促進鋰硫電池的實際應用具有重要意義。
References
[1] MANTHIRAM A, FU Y, CHUNG S H, ZU C, SU Y S. Chem. Rev. , 2014, 114(23): 11751-11787.
[2] BRUCE P G, FREUNBERGER S A, HARDWICK L J, TARASCON J M. Nat. Mater. , 2011, 11(1): 19-29.
[3] ZHOU S, SHI J, LIU S, LI G, PEI F, CHEN Y, DENG J, ZHENG Q, LI J, ZHAO C, HWANG I, SUN C J, LIU Y, DENG Y,HUANG L, QIAO Y, XU G L, CHEN J F, AMINE K, SUN S G, LIAO H G. Nature, 2023, 621(7977): 75-81.
[4] LIU R, WEI Z, PENG L, ZHANG L, ZOHAR A, SCHOEPPNER R, WANG P, WAN C, ZHU D, LIU H, WANG Z,TOLBERT S H, DUNN B, HUANG Y, SAUTET P, DUAN X. Nature, 2024, 626(7997): 98-104.
[5] YAO W, LIAO K, LAI T, SUL H, MANTHIRAM A. Chem. Rev. , 2024, 124(8): 4395-5118.
[6] FANG R, ZHAO S, SUN Z, WANG D W, CHENG H M, LI F. Adv. Mater. , 2017, 29(48): 1606823.
[7] KANG H, SUN Y. Adv. Funct. Mater. , 2016, 26(8): 1225-1232.
[8] ZHAO C, XU G L, YU Z, ZHANG L, HWANG I, MO Y X, REN Y, CHENG L, SUN C J, REN Y, ZUO X, LI J T, SUN S G,AMINE K, ZHAO T. Nat. Nanotechnol. , 2021, 16(2): 166-173.
[9] LIU G X, TIAN J X, WAN J, LI Y, SHEN Z Z, CHEN W P, ZHAO Y, WANG F, LIU B, XIN S, GUO Y G, WEN R. Angew.Chem. Int. Ed. , 2022, 61(52): e202212744.
[10] MAO Y, LI G, GUO Y, LI Z, LIANG C, PENG X, LIN Z. Nat. Commun. , 2017, 8(1): 14628.
[11] ZHONG Y, WANG Q, BAK S M, HWANG S, DU Y, WANG H. J. Am. Chem. Soc. , 2023, 145(13): 7390-7396.
[12] LUO Y, FANG Z, DUAN S, WU H, LIU H, ZHAO Y, WANG K, LI Q, FAN S, ZHENG Z, DUAN W, ZHANG Y, WANG J.Angew. Chem. Int. Ed. , 2023, 62(11): e202215802.
[13] SONG Y, CAI W, KONG L, CAI J, ZHANG Q, SUN J. Adv. Energy Mater. , 2019, 10(11): 1901075.
[14] WU M, CUI Y, BHARGAV A, LOSOVYJ Y, SIEGEL A, AGARWAL M, MA Y, FU Y. Angew. Chem. Int. Ed. , 2016,55(34): 10027-10031.
[15] HUA W, YANG Z, NIE H, LI Z, YANG J, GUO Z, RUAN C, CHEN X, HUANG S. ACS Nano, 2017, 11(2): 2209-2218.
[16] TSAO Y, LEE M, MILLER E C, GAO G, PARK J, CHEN S, KATSUMATA T, TRAN H, WANG L W, TONEY M F, CUI Y,BAO Z. Joule, 2019, 3(3): 872-884.
[17] HUA W, LI H, PEI C, XIA J, SUN Y, ZHANG C, LV W, TAO Y, JIAO Y, ZHANG B, QIAO, WAN Y, YANG Q H. Adv.Mater. , 2021, 33(38): e2101006.
[18] ZHANG Z, LUO D, LI G, GAO R, LI M, LI S, ZHAO L, DOU H, WEN G, SY S, HU Y, LI J, YU A, CHEN Z. Matter, 2020,3(3): 920-934.
[19] GUO W, ZHANG W, SI Y, WANG D, FU Y, MANTHIRAM A. Nat. Commun. , 2021, 12(1): 3031.
[20] LI Z, SAMI I, YANG J, LI J, KUMAR R V, CHHOWALLA M. Nat. Energy, 2023, 8(1): 84-93.
[21] LIU Zhen-Bang, MA Ying-Ming, HAN Dong-Xue, DONG Xian-Dui, NIU Li, BAO Yu. Chin. J. Anal. Chem. , 2018, 46(8):1171-1177.
劉振邦, 馬英明, 韓冬雪, 董獻堆, 牛利, 包宇. 分析化學, 2018, 46(8): 1171-1177.
[22] LIAO Yu-Zhi, SI Shi-Hui, CHEN Jin-Hua, LU Yang, DU Ming. Chin. J. Anal. Chem. , 2019, 47(7): 992-997.
廖玉枝, 司士輝, 陳金華, 盧陽, 杜明. 分析化學, 2019, 47(7): 992-997.
[23] PENG L, WEI Z, WAN C, LI J, CHEN Z, ZHU D, BAUMANN D, LIU H, ALLEN C S, XU X, KIRKLAND A I, SHAKIR I,ALMUTAIRI Z, TOLBERT S, DUNN B, HUANG Y, SAUTET P, DUAN X. Nat. Catal. , 2020, 3(9): 762-770.
[24] XU N, QIAN T, LIU X, LIU J, CHEN Y, YAN C. Nano Lett. , 2017, 17(1): 538-543.
國家自然科學基金項目(Nos. 22374141, 22134006, U2241287)資助。
猜你喜歡
現代養生·下半月(2016年5期)2017-01-09 10:12:30
中國當代醫藥(2016年30期)2017-01-07 00:55:28
中國當代醫藥(2016年30期)2017-01-07 00:49:32
中國實用醫藥(2016年30期)2016-12-28 22:59:47
中國當代醫藥(2016年28期)2016-12-20 18:12:21
中國醫藥導報(2016年25期)2016-11-30 07:20:06
中外醫學研究(2016年28期)2016-11-28 07:12:34
中國實用醫藥(2016年26期)2016-11-07 13:49:05
中國實用醫藥(2016年24期)2016-10-17 05:10:07
中國實用醫藥(2016年24期)2016-10-17 04:52:27
