改進QuEChERS/超高效液相色譜-串聯三重四極桿質譜法快速檢測茶葉中百草枯和敵草快殘留
2024-11-24 00:00:00唐燕花高英楠陳江艷郭心怡李紹涌李悅周振高偉
分析化學 2024年10期


關鍵詞 QuEChERS;超高效液相色譜-串聯三重四極桿質譜法;茶葉;百草枯;敵草快
百草枯和敵草快是廣譜性、接觸性的除草劑,由于成本較低被廣泛用于茶園中以清除雜草[1]。百草枯對人畜毒性極強,早期中毒可引起急性肺損傷或急性呼吸窘迫綜合征等嚴重病癥,多數誤食者死于呼吸衰竭[2-3]。鑒于百草枯的高毒性,我國于2016 年7 月起禁止銷售和使用該藥物,隨后,百草枯的有效替代品敵草快的市場銷量呈現增長趨勢[4]。敵草快是一種中等毒性的除草劑,其毒性低于百草枯,但其致毒機理與百草枯相似,并且兩者分子結構均穩定,難以降解,易殘留在茶葉等農作物上,可通過食物鏈進入人畜體內,對人畜的健康與生命安全構成了嚴重威脅[5]。因此,建立一種茶葉中百草枯和敵草快殘留量的簡單、快速和高效的檢測方法十分重要。
目前,百草枯和敵草快的檢測涉及環境基質(如水體[6])、生物基質(如尿液和人血漿[7-8]等)以及食品基質(如小麥[9]、蘋果[10]和茶葉[11]等),檢測方法包括伏安分析法[12]、分光光度法[13]、免疫熒光法[6]、氣相色譜法(GC)和氣相色譜-質譜法(GC-MS)[14-15]、高效液相色譜法(HPLC)或高效液相色譜-串聯質譜法(HPLC-MS/MS)等[16-18],其中, HPLC-MS/MS 因具有較好的選擇性和靈敏度,廣泛應用于茶葉中百草枯和敵草快的分析測定。通常,復雜基質樣品的檢測需采用適當的前處理技術進行富集和凈化。固相萃取是一種常用的前處理技術,可有效地去除雜質,提高檢測的靈敏度和準確度,但常規固相萃取過程繁瑣且成本較高,不適于茶葉中百草枯和敵草快的快速測定。QuEChERS 作為常用的農殘檢測前處理手段,因具有操作簡單、快速、安全和成本低等優點而被廣泛采用[19-20]。
本研究基于改進的QuEChERS 法結合超高效液相色譜-串聯三重四極桿質譜技術,建立了一種可快速、準確測定茶葉中百草枯和敵草快殘留量的檢測方法,以期為植物源性的食品中除草劑殘留量的檢測提供技術補充。
1 實驗部分
1.1 儀器與試劑
LC-TQ 5200 超高效液相色譜-三重四極桿質譜聯用儀(廣州禾信儀器股份有限公司);AUW220 型萬分之一分析天平(德國Sartorius 公司);TG19 型高速離心機(上海盧湘儀公司);XH-B 型渦旋混合器(江蘇天翎公司);Millipore 超純水處理系統(德國Merck 公司);AK-030SD 超聲波清洗器(深圳鈺潔公司)。
二氯百草枯和敵草快二溴鹽標準溶液(1000 μg/mL,天津阿爾塔有限公司);甲酸銨(色譜純,上海安譜公司);甲醇和乙腈(質譜純,上海安譜公司);甲酸(色譜純,美國Aladdin 試劑公司);C18 和N-丙基乙二胺(PSA)(40~60 μm,深圳逗點生物技術有限公司)。碧螺春、烏龍茶、黑茶和紅茶均購于當地超市;茶葉陽性樣品來自國家茶葉質量檢測監督中心(福建)。
1.2 實驗方法
1.2.1 百草枯和敵草快混合標準溶液的配制
分別移取適量的1000 μg/mL 二氯百草枯和敵草快二溴鹽標準溶液于10 mL 棕色容量瓶中,用甲醇稀釋并定容,配制成10 μg/mL 百草枯和敵草快的混合標準儲備液,于?18 ℃下保存。
移取百草枯和敵草快混合標準儲備液,用0.1%甲酸作為溶劑分別配制5、10、100 和1000 μg/L 的百草枯和敵草快混合標準工作液,于4 ℃下保存。
1.2.2 樣品前處理
稱取磨碎后的茶粉樣品0.5 g 于15 mL 塑料離心管中(茶葉樣品磨碎方式參考GB/T 8303—2013[21]),加入5 mL 乙腈-0.1%甲酸(3∶7, V/V)溶液,渦旋振蕩1 min 充分混勻,超聲提取15 min,以10000 r/min 離心5 min,準確移取3 mL 上清液至15 mL 塑料離心管中。向上清液中加入凈化劑(400 mg C18 和400 mgPSA),渦旋振蕩混勻1 min, 10000 r/min 離心5 min,取上清液,過0.22 μm 濾膜過濾,待測。
1.2.3 色譜條件
采用親水型HILIC 色譜柱(100 mm×2.1 mm, 1.8 μm,上海安譜科技公司);柱溫為30 ℃;進樣體積為10 μL;流速為0.3 mL/min;流動相A 為0.1%甲酸(含5 mmol/L 甲酸銨), B 為乙腈;等度洗脫程序:0~4 min, 50% A/50% B。
1.2.4 質譜條件
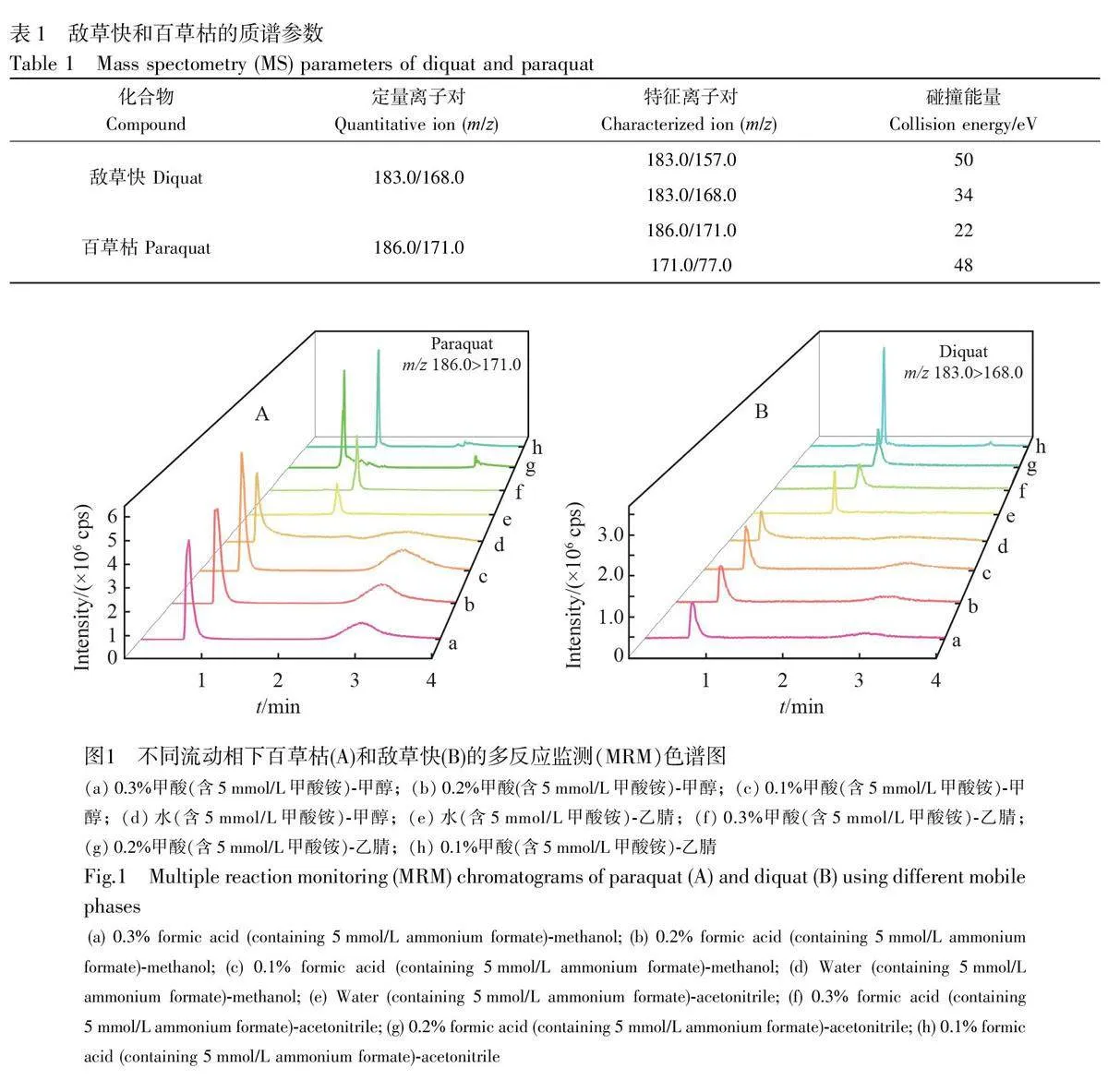
電噴霧電離源(ESI+);多反應監測模式(MRM);干燥氣加熱溫度為500 ℃;噴霧電壓為5500 V;霧化氣為70 psi(1 psi=6.89 kp);干燥氣流速為8 L/min;反吹氣流速為3 L/min;碰撞氣流速為0.4 mL/min;氣簾盤高壓為800 V;真空接口電壓為70 V;檢測器電壓為2300 V;碰撞池入口電壓為30 V;碰撞池出口電壓為1 V。
2 結果與討論
2.2 色譜條件的優化
2.2.1 流動相的選擇
流動相對色譜分析至關重要,流動相組成會影響分析方法的回收率、色譜峰形、離子化效率和靈敏度,選擇合適的流動相可提高回收率、改善峰形和消除樣品基質干擾等[24]。本研究考察了甲醇和乙腈兩種有機相與不同pH 值的水相流動相對百草枯和敵草快的響應和峰形的影響,比較了不同流動相體系對待測物響應和峰形的影響。如圖1 所示,在水相中加入甲酸,能夠促進目標物電離,提高待測物響應;與甲醇相比,乙腈作為有機相時,兩種目標物峰形更尖銳,拖尾和鼓包現象均有所改善。以0.1%甲酸(含5 mmol/L 甲酸銨)-乙腈作為流動相時,可有效改善峰形、提高離子化效率、提升靈敏度。因此,本研究選擇0.1%甲酸(含5 mmol/L 甲酸銨)-乙腈作為流動相進行分析。
2.2.2 色譜柱的選擇
百草枯和敵草快屬于季銨鹽化合物,為極性物質,易溶于水。高極性物質對反相色譜柱(C18 和T3)固定相的親和力較低,保留效果不理想,并且峰形不佳[1]。親水型HILIC 色譜柱采用強極性物質作為固定相,能夠有效改善極性物質的保留效果和峰形,故本研究選擇親水型HILIC 色譜柱進行分析。
2.3 提取方法的優化
2.3.1 提取溶劑的優化
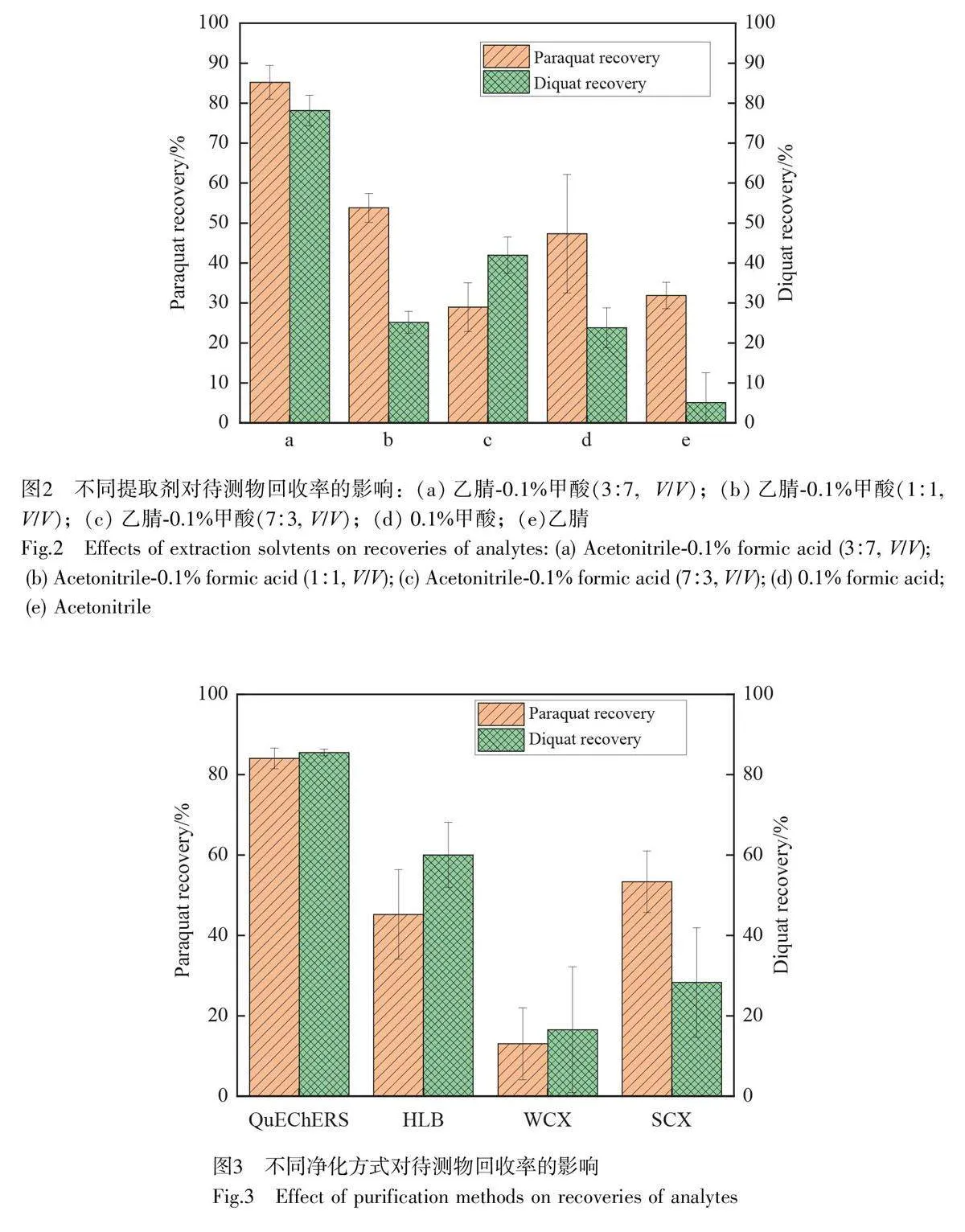
考察了不同提取劑對茶葉加標樣品中百草枯和敵草快提取效果的影響,選取0.1%甲酸、純乙腈、乙腈-0.1%甲酸(3∶7、1∶1、7∶3, V/V)5 種提取劑進行實驗,以回收率作為評價指標。如圖2 所示,百草枯和敵草快的水溶性極強,采用單一的有機溶劑提取溶時,目標物的回收率低于50%;百草枯和敵草快均為堿性物質,酸性溶液能提高其溶解性,采用0.1%甲酸提取時,兩者回收率均有所提升;在0.1%甲酸中加入適量極性溶劑乙腈,可更好地溶解基質中的脂肪酸和氨基酸等物質,兩種目標物的回收率均進一步提高。研究結果表明,采用乙腈-0.1%甲酸(3∶7, V/V)提取時,百草枯和敵草快的回收率分別可達到85.21%和78.14%。因此,本方法選擇乙腈-0.1%甲酸(3∶7, V/V)作為提取劑。
2.3.2 凈化方式的選擇
食品檢測前處理常用的凈化方式有固相萃取法和QuEChERS 法。固相萃取法過程繁瑣且成本較高,而QuEChERS 法操作簡單、高效快速、安全可靠且成本較低,在食品檢測領域備受關注[25]。百草枯和敵草快的pKa 都大于10,在水溶液中呈堿性,因此,本研究探究了SCX(強陽離子交換柱)、WCX(弱陽離子交換柱)和HLB(親水性反相吸附柱)3 種陽離子交換柱對茶葉加標樣品中待測物的提取效果。如圖3所示,采用SCX 和HLB 凈化時,茶葉中的茶多酚、咖啡因和色素等成分無法有效去除,提取液渾濁,目標物的回收率均低于60%;采用WCX 凈化時,茶葉提取液出現絮狀沉淀物,可能是采用NaOH 溶液調節pH 值時,在離子效應、電解質作用和共沉效應等共同作用下,茶多酚、氨基酸和糖類等聚合形成絡合物而沉淀,影響了過柱效果,導致回收率低于20%。采用QuEChERS 法進行凈化時,常用的凈化材料有無水MgSO4、C18 和PSA,其中,無水MgSO4 可除去水分,但兩種目標物為水溶性物質,無水MgSO4 將導致目標物損失。本研究選用C18 和PSA 作為凈化劑,乙腈-0.1%甲酸3∶7(V/V)作為提取溶劑,得到的提取液澄清,回收率大于80%且處理速度快,所用耗材成本低。
2.3.3 C18 和PSA用量的優化
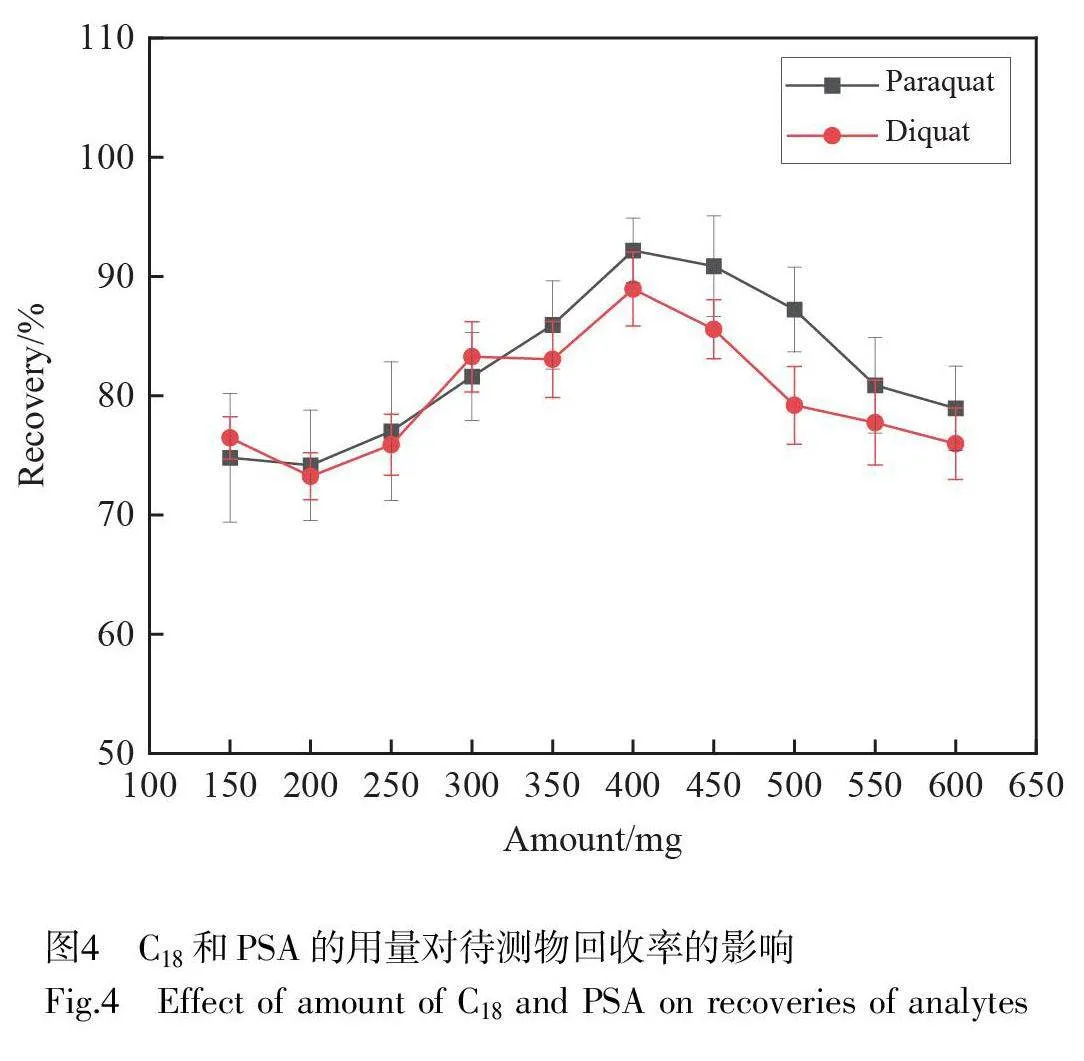
C18 可去除一些非極性干擾物,如油脂和固醇等;PSA 能去除極性干擾物,如有機酸、糖類和色素等。本研究采用C18 和PSA 對茶葉進行凈化,并考察了不同C18 和PSA 用量(150、200、250、300、350、400、450、500、550 和600 mg)對百草枯和敵草快回收率的影響。如圖4 所示,當C18 和PSA 用量為400 mg 時,百草枯和敵草快的回收率均大于80%。因此,本研究中C18 和PSA 的用量為400 mg。
2.4 基質效應
采用空白碧螺春、白茶、紅茶和烏龍茶基質考察本方法的基質效應(Matrix effect, ME),分別用4 種基質空白溶液和乙腈-0.1%甲酸(3∶7, V/V)作為溶劑配制得到系列濃度溶液。ME 的計算公式如下:
其中, A 和B 分別為茶葉基質溶液曲線和乙腈-0.1%甲酸溶劑曲線的斜率。
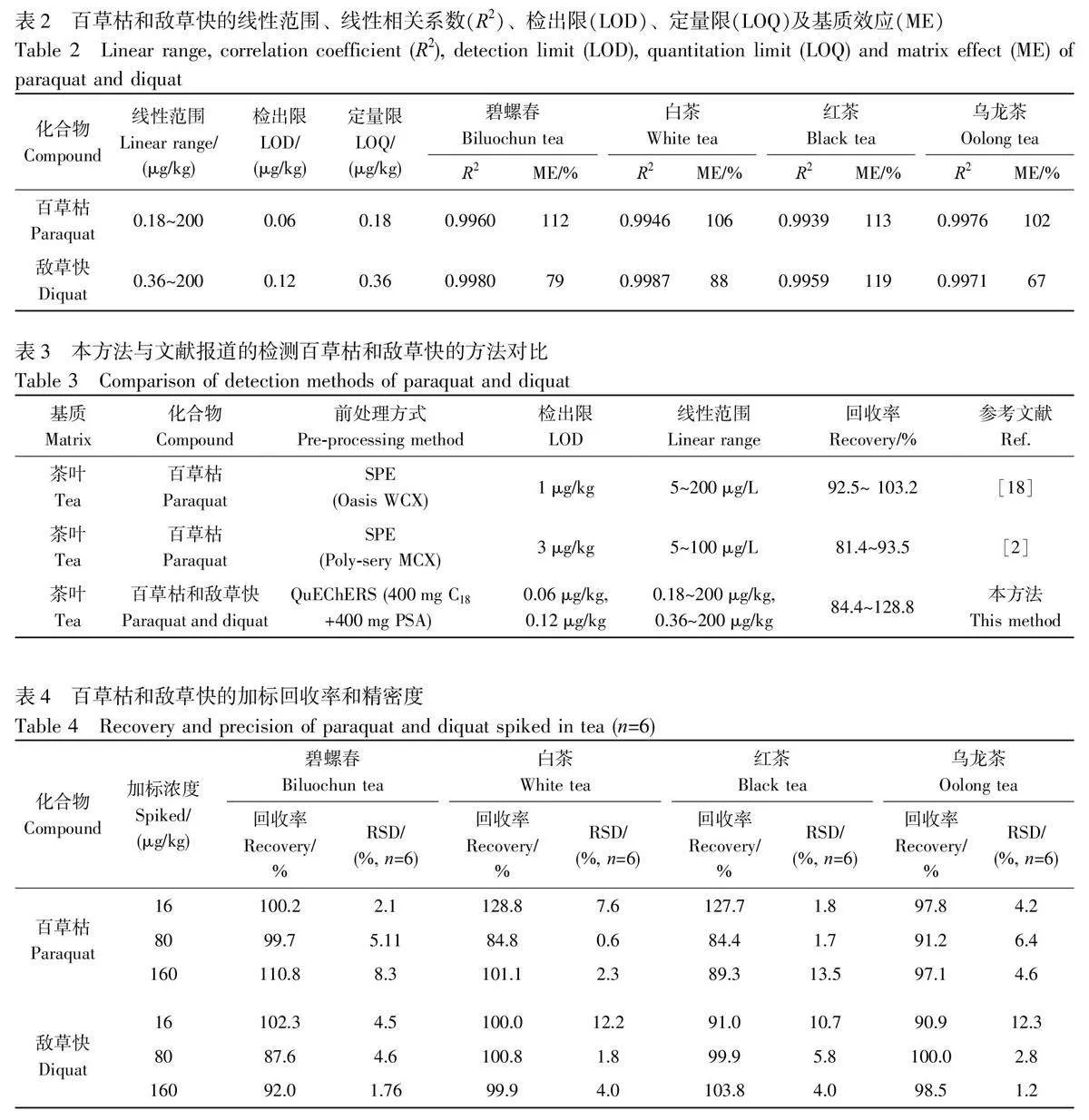
通常,當ME 為85%~120%時,表明基質效應影響可忽略;當MEgt;120%或MElt;85%時,表明存在基質效應增強或基質效應抑制,需采用適當的方法進行校準。敵草快和百草枯的基質效應數據見表2,4 種茶葉對百草枯的基質效應影響較弱,而對敵草快存在基質效應增強和抑制的影響。為保證實驗結果的準確性和可靠性,后續實驗中均采用基質匹配標準溶液進行定量分析。
2.5 方法學驗證
2.5.1 線性關系和方法檢出限
采用碧螺春、白茶、紅茶和烏龍茶4 種茶葉空白基質溶液分別配制百草枯和敵草快的系列濃度工作溶液,以待測化合物的質量濃度(x)為橫坐標、定量離子對的色譜峰面積(y)為縱坐標,進行線性回歸擬合,得到線性方程和相關系數;分別以離子對的信噪比≥3 和≥10 時的樣品濃度作為檢出限(LOD)和定量限(LOQ)。4 種茶葉中的百草枯和敵草快的定量分析評價指標見表2,兩種待測物相關系數均大于0.99,線性關系良好。
與文獻報道的除草劑尤其是百草枯和敵草快的分析方法相比(表3),本方法具有較好的靈敏度、較寬的線性范圍以及良好的回收率,并且操作簡單、分析速度快,為百草枯和敵草快的檢測提供了一種方法補充。
2.5.2 加標回收率和精密度
選取空白碧螺春、白茶、紅茶和烏龍茶樣品,對其進行不同濃度水平的加標回收實驗。采用標準工作曲線外標法進行定量分析,每個加標濃度水平平行測試6 次,加標回收率如表4 所示,百草枯和敵草快的平均回收率分別為84.4%~128.8%和87.6%~103.8%,相對標準偏差(RSD)分別為0.6%~13.5%和1.2%~12.3%,說明本方法的準確度和特密度良好。
2.6 實際樣品分析
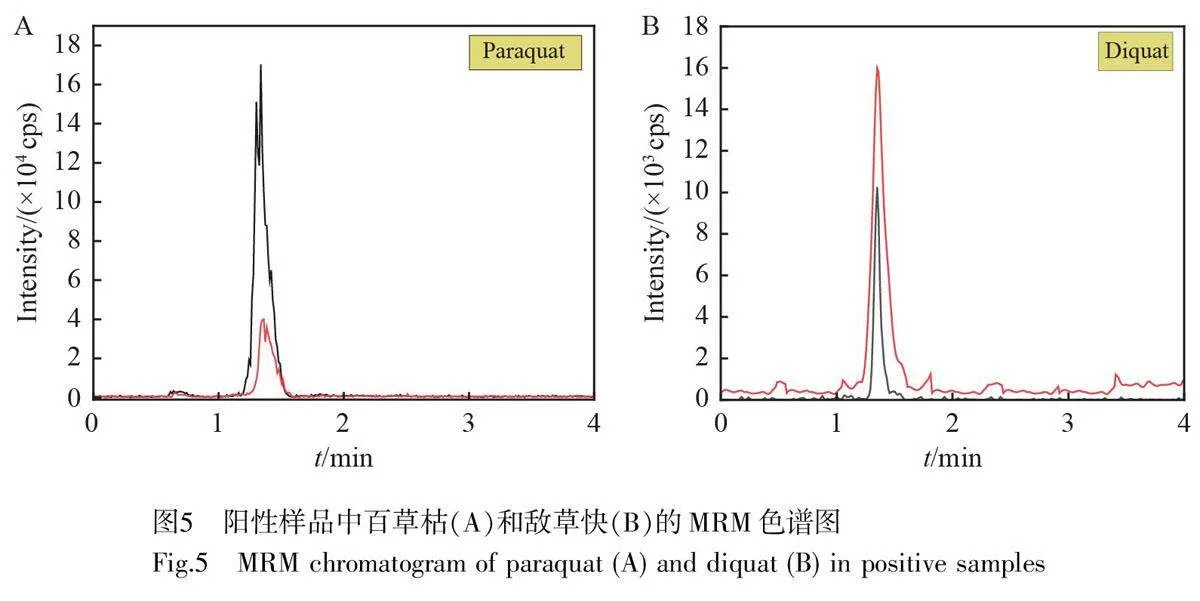
采用本方法對碧螺春、白茶、紅茶和烏龍茶4 種茶葉樣品以及國家茶葉質量檢測監督中心(福建)提供的茶葉陽性樣品中的百草枯和敵草快的殘留量進行檢測,在4 種茶葉樣品均未檢出百草枯和敵草快,茶葉陽性樣品中檢出百草枯和敵草快的含量分別為9.0 μg/kg 和9.5 μg/kg,樣品檢測的MRM 色譜圖如圖5所示。
3 結論
建立了一種茶葉中百草枯和敵草快殘留量的快速檢測方法, 4 種茶葉基質中百草枯和敵草的線性關系良好, R2gt;0.99,方法檢出限分別為0.06 和0.12 μg/kg,定量限分別為0.18 和0.36 μg/kg,平均加標回收率為84.4%~128.8%, RSD 為0.6%~13.5%。本方法簡單、快速、準確,適用于茶葉樣品中百草枯和敵草快的檢測,為植物源性食品中除草劑殘留量的快速檢測分析提供了方法補充。
