超高效液相色譜-串聯質譜法同時測定當歸9種真菌毒素
2024-11-07 00:00:00李玉芳焦潔黃錚肖鋒黃遠飛王青柳利龍張環
甘肅農業科技 2024年10期

摘要:通過優化液相色譜條件和質譜條件等,建立超高效液相色譜-串聯質譜法(UPLC-MS/MS)測定當歸中藥材中9種真菌毒素的檢測方法。樣品經70%甲醇溶液超聲提取,經固相萃取柱凈化;采用流動相A:2 mmol/L甲酸銨0.1%甲酸水溶液;流動相B:甲醇,經C18色譜柱分離后注入質譜儀,電噴霧電離源(ESI)和多反應監測模式(MRM)進行檢測,基質外標法定量。經方法學驗證,9種真菌毒素標準曲線線性關系良好(R2 > 0.995 0),方法的檢出限0.04~1.43 μg/kg,定量限0.12~4.75 μg/kg,高、中、低3個濃度加標回收率為77.8%~113.6%,測定結果的相對標準偏差為0.2%~17.7%。該檢測方法應用于72批當歸樣品9種真菌毒素同時檢測,2批當歸樣品檢出真菌毒素,檢出率為2.8%,樣品中主要的污染真菌毒素為黃曲霉毒素B1(AFTB1)、黃曲霉毒素B2(AFTB2)、黃曲霉毒素G1(AFTG1)。本研究建立的方法具有預處理簡便、經濟等優點,適用于當歸樣品9種真菌毒素的快速檢測。
關鍵詞:當歸;中藥材;真菌毒素;超高效液相色譜-串聯質譜法;方法驗證
中圖分類號:S567.23 文獻標志碼:A 文章編號:2097-2172(2024)10-0974-07
doi:10.3969/j.issn.2097-2172.2024.10.017
Simultaneous Determination of 9 Mycotoxins in Angelica sinensis by Ultra High Performance Liquid Chromatography-tandem Mass Spectrometry
LI Yufang 1, 2, JIAO Jie 1, 2, HUANG Zheng 1, 2, XIAO Feng 3, HUANG Yuanfei 3,
WANG Qing 1, 2, LIU Lilong 4, ZHANG Huan 1, 2
(1. Institute of Animal Husbandry, Pasture and Green Agriculture, Gansu Academy of Agricultural Sciences, Lanzhou Gansu 730070, China; 2. Institute of Agricultural Quality Standards and Testing Technology, Gansu Academy of Agricultural Sciences, Lanzhou
Gansu 730070, China; 3. Wenshan Sanqi Digital Materia Medica Inspection Centre Co., Ltd., Wenshan Yunnan 663000, China;
4. Wheat Research Institute, Gansu Academy of Agricultural Sciences, Lanzhou Gansu 730070, China)
Abstract: An ultra-high performance liquid chromatography-tandem mass spectrometry(UPLC-MS/MS) method for the determination of 9 mycotoxins in Angelica sinensis was established by optimizing the conditions of liquid chromatography and mass spectrometry. The samples were extracted by ultrasonic extraction of 70% methanol solution and purified by solid phase extraction column. Using mobile phase A, i.e., 0.1% formic acid solution(containing 2 mmol/L ammonium formate), and mobile phase B, i.e., methanol, samples were separated by a C18 column and injected into a mass spectrometer, detected by ESI and multiple reaction monitoring mode (MRM), and quantified by matrix external standard. The linear relationship between the standard curves of the nine mycotoxins was good (R2 > 0.995). The detection limits were 0.04 to1.43 μg/kg, and the quantification limits were 0.12 to 4.75 μg/kg. The recoveries of high, medium and low concentrations were 77.8% to 113.6%. The relative standard deviation of the results was 0.2% to17.7%. The method was applied to the simultaneous detection of mycotoxins in 72 batches of Angelica sinensis samples. Mycotoxins were detected in 2 batches of Angelica sinensis samples, with a detection rate of 2.8%. The main mycotoxins in Angelica sinensis samples were AFTB1, AFTB2 and AFTG1. The method established in this study has the advantages of simple pretreatment and economy, and is suitable for the rapid detection of 9 mycotoxins in Angelica sinensis samples.
Key words: Angelica sinensis; Chinese medicinal material; Mycotoxin; Ultra high performance liquid chromatography-tandem mass spectrometry; Method validation
收稿日期:2024 - 08 - 16
基金項目:甘肅省農業科學院農業科技創新專項(2020GAAS15);甘肅省自然科學基金計劃項目(22JR5RA764)。
作者簡介:李玉芳(1964 — ),女,甘肅武威人,副研究員,主要從事農產品質量安全控制與檢測技術研究工作。Email: 1292153904@qq.com。
通信作者:焦 潔(1983 — ),女,甘肅通渭人,主要從事農產品質量安全檢測技術工作。Email: 14199634@qq.com。
當歸為傘形科植物Angelica sinensis(Oliv.)Diels的干燥根,具有較高的藥用價值,故有“十方九歸”的美譽[1 ]。當歸具有補血活血、潤腸通便、調經止痛等多重功效,能夠有效促進血液循環,緩解疼痛,并對腸道功能有良好的調節作用[2 ]。現代藥理研究表明,當歸多糖、阿魏酸和當歸揮發油是當歸發揮造血、免疫調節、抗腫瘤、肝保護、神經保護等作用的主要標志化合物。這些化合物的藥理作用機制已經得到充分研究和證實,并在功能食品開發、藥物創新研發方面具有較大潛力[3 - 4 ]。當歸在甘肅省種植歷史久遠、品質卓越,近年來種植面積穩定在2.3萬hm2以上,當歸產業發展占全國出口總量的90%以上,形成了較為完善的產業鏈和銷售渠道。當歸在種植、采集、加工、儲藏及運輸的多個環節極易遭受真菌污染,真菌在當歸內部滋生繁衍,不僅可能破壞其珍貴的功效成分,還可能產生多種真菌毒素污染,進而對中藥的整體質量和療效造成嚴重影響[5 - 7 ]。為有效管控中藥真菌毒素污染,部分國家和地區對中藥材真菌毒素的含量實施了強制性規定,并設立了嚴格的限量標準[8 - 9 ]。
近年來,超高效液相色譜-串聯質譜法(UPLC-MS/MS)在中藥材真菌毒素檢測技術領域取得了顯著進展,并得到了廣泛應用[10 - 14 ]。該方法不僅提升了檢測的靈敏度和準確性,還顯著提高了分析效率,滿足現代社會對中藥材安全快速、準確檢測的需求。本研究同時提取當歸樣品中9種真菌毒素,采用固相萃取柱富集凈化,正離子和負離子切換模式和多反應監測(MRM)模式進行檢測,建立了UPLC-MS/MS同時檢測當歸中9種真菌毒素的方法,以期為客觀評價甘肅省當歸藥材的安全性提供技術支持和參考。
1 材料與方法
1.1 儀器與試劑
1.1.1 儀器 液相色譜-串聯質譜儀(1290系列液相色譜儀、6470系列三重四極桿串聯質譜儀,美國Agilent公司);電噴霧電離源(Electrospray ionization,ESI);渦旋儀(XW-80A,上海瀘西分析儀器廠);純水系統(PALL/CascadaI,美國PALL公司);氮吹儀(HGC-24A,天津市恒奧科技發展有限公司);超聲波清洗器(KQ5200B,昆山市超聲儀器有限公司);HLB固相萃取柱(TC-MT3000,德國拜發公司);HLB固相萃取柱[3 mL/200 mg,微純生物科技(廣州)有限公司];分析天平(AS220-R2,蘇州培科實驗室儀器科技有限公司)。
1.1.2 試劑 黃曲霉毒素B1(AFTB1)、黃曲霉毒素B2(AFTB2)、黃曲霉毒素G1(AFTG1)、黃曲霉毒素G2(AFTG2)均購自美國Sigma-Aldrich公司,質量濃度分別為1.0、0.3、1.0、0.3 μg/mL;脫氧雪腐鐮刀菌烯醇(DON,100 μg/mL)購自壇墨質檢科技股份有限公司;伏馬毒素B1(FB1,100μg/mL)、伏馬毒素B2(FB2,100 μg/mL)、赭曲霉毒素A(OTA, 100 μg/mL)、玉米赤霉烯酮(ZEN,100 μg/mL)均購自天津阿爾塔科技有限公司。甲醇(色譜純,天津市康科德科技有限公司);甲酸(分析純,天津市風船化學試劑科技有限公司);乙酸銨(分析純,西隴科學股份有限公司)。
1.1.3 樣品 來自甘肅省當歸主產區岷縣、隴西縣、卓尼縣、漳縣、宕昌縣、臨潭縣等地的農戶、生產合作社、生產企業和中藥材市場采集的當歸樣品,共計72批次。
1.2 試驗方法
1.2.1 樣品前處理 取粉碎后過二號篩(850±29 μm)的當歸粉末5.00 g,加入70%甲醇溶液50.0 mL,超聲處理30 min后離心。吸取上清液5.00 mL,緩緩通過已提前依次用甲醇和3 mL水預活化的HLB柱,直至柱中空氣通過,收集洗脫液。再以3 mL甲醇進行第二次洗脫,并收集洗脫液,將2次洗脫所得的液體合并,于40 ℃下利用氮氣將其緩慢吹至剩余2 mL,用微孔濾膜(0.22 μm)過濾后得到上機液。取未經真菌毒素污染的當歸樣品,按供試樣品前處理方法制備當歸空白基質溶液。
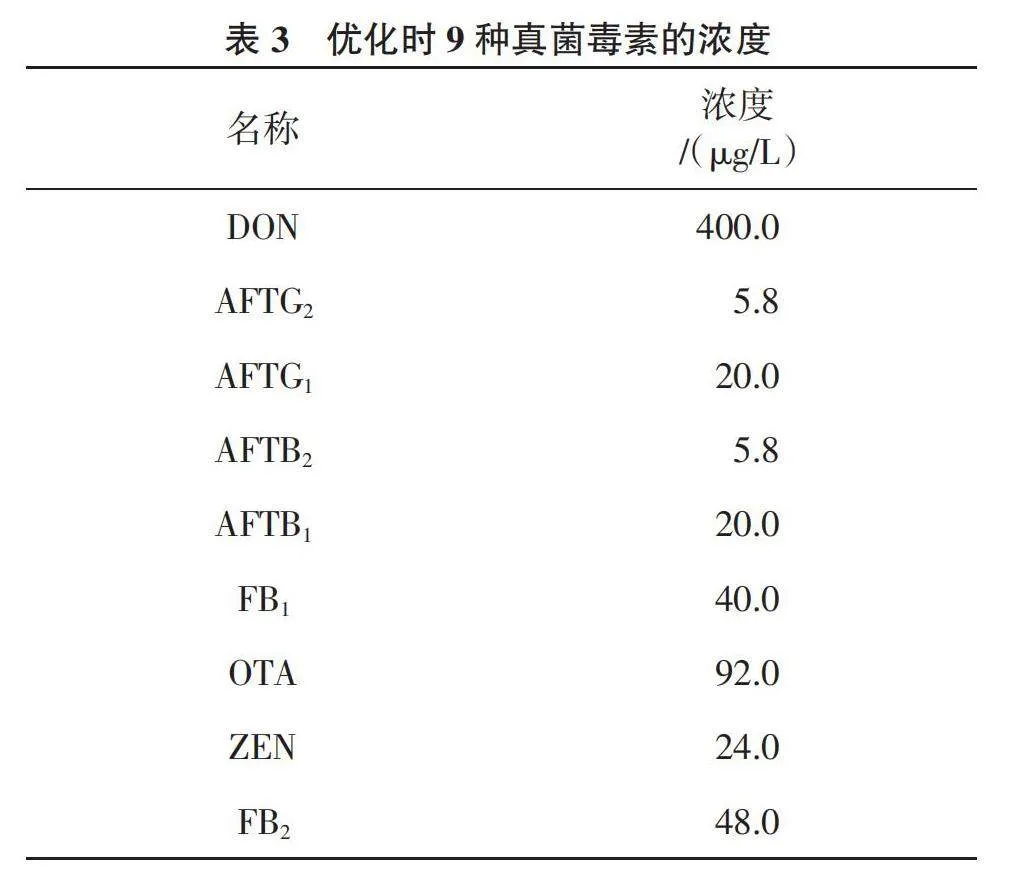
1.2.2 標準溶液 對照品儲備溶液的制備:分別精密吸取AFTB1、AFTB2、AFTG1、AFTG2、DON、FB1、FB2、OTA、ZEN標準溶液各1.0 mL至10 mL容量瓶中,用甲醇稀釋至刻度并混勻,置于-20 ℃避光條件下保存。混合對照品溶液的制備:分別精密量取AFTB1 10.00 mL、AFTB2 10.00 mL、AFTG1 10.00 mL、AFTG2 10.00 mL、DON 0.10 mL、FB1 0.10 mL、FB2 0.10 mL、OTA 0.20 mL、ZEN 0.05 mL至20 mL容量瓶中,加入甲醇稀釋至刻度后混勻。混合標準溶液中各組分濃度為AFTB1 25.00 ng/mL、AFTB2 7.25 ng/mL、AFTG1 25.00 ng/mL、AFTG2 7.25 ng/mL、DON 500.00 ng/mL、FB1 50.00 ng/mL、FB2 60.00 ng/mL、OTA1 15.00 ng/mL、ZEN 30.00 ng/mL,-20 ℃避光條件下保存。混合對照溶液標準曲線的制備:分別精密量取混合對照品溶液0.16、0.20、0.32、0.40、0.80、1.00 mL,氮氣吹干,加當歸空白基質溶液1.0 mL,混勻后即得AFTG1、AFTB1(4、5、8、10、20、25 ng/mL),AFTG2、AFTB2(1.16、1.45、2.32、2.90、5.80、7.25 ng/mL),DON(80、100、160、200、400、500 ng/mL),FB1(8、10、16、20、40、50 ng/mL),FB2(9.6、12.0、19.2、24.0、48.0、60.0 ng/mL),OTA(18.4、23.0、36.8、46.0、92.0、115.0 ng/mL),ZEN(4.8、6.0、9.6、12.0、24.0、30.0 ng/mL)的系列標準曲線。
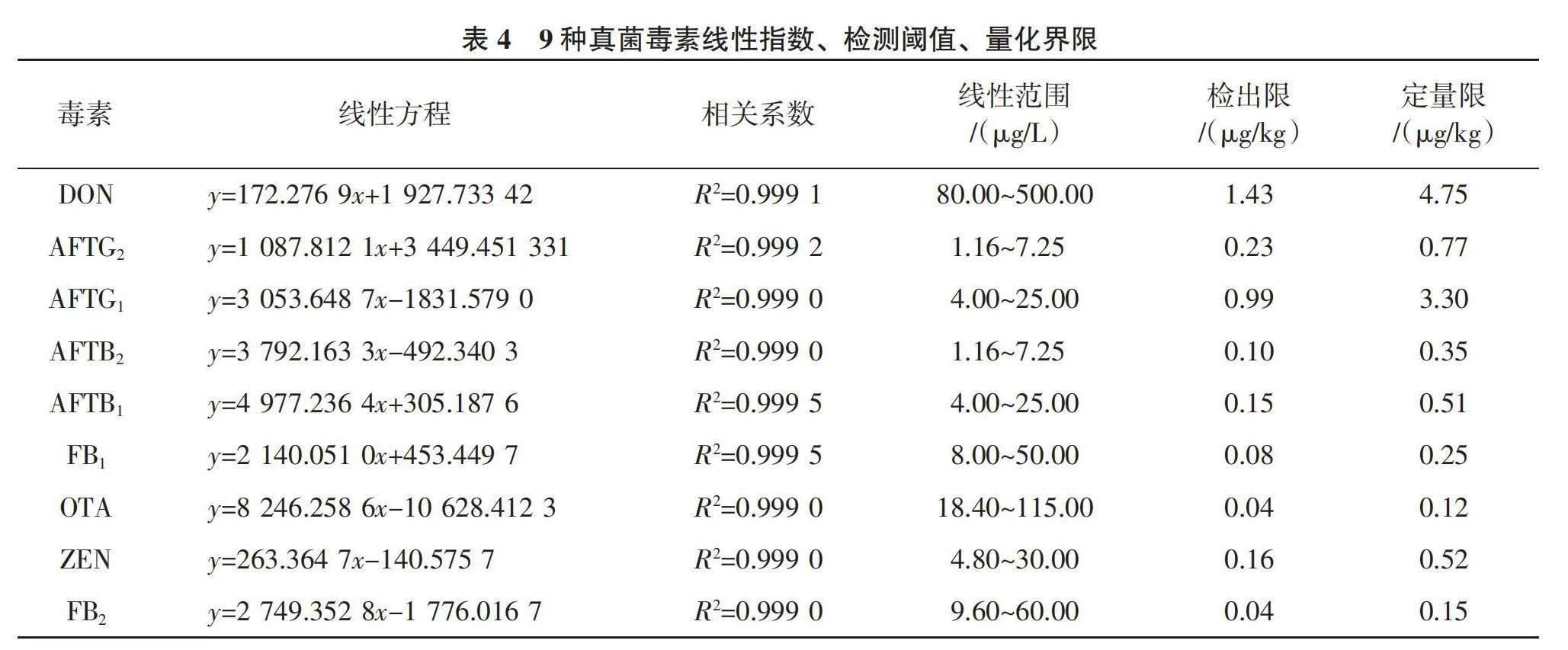
1.2.3 液相色譜條件 參考劉笑笑等[15 ]的方法,液相色譜柱為Hypersil GOLDTMaQ色譜柱;流動相A:2 mmol/L甲酸銨0.1%甲酸水溶液;流動相B:甲醇;流速:0.3 mL/min,進樣體積:5 μL;柱溫:25 ℃;流動相梯度洗脫程序見表1。
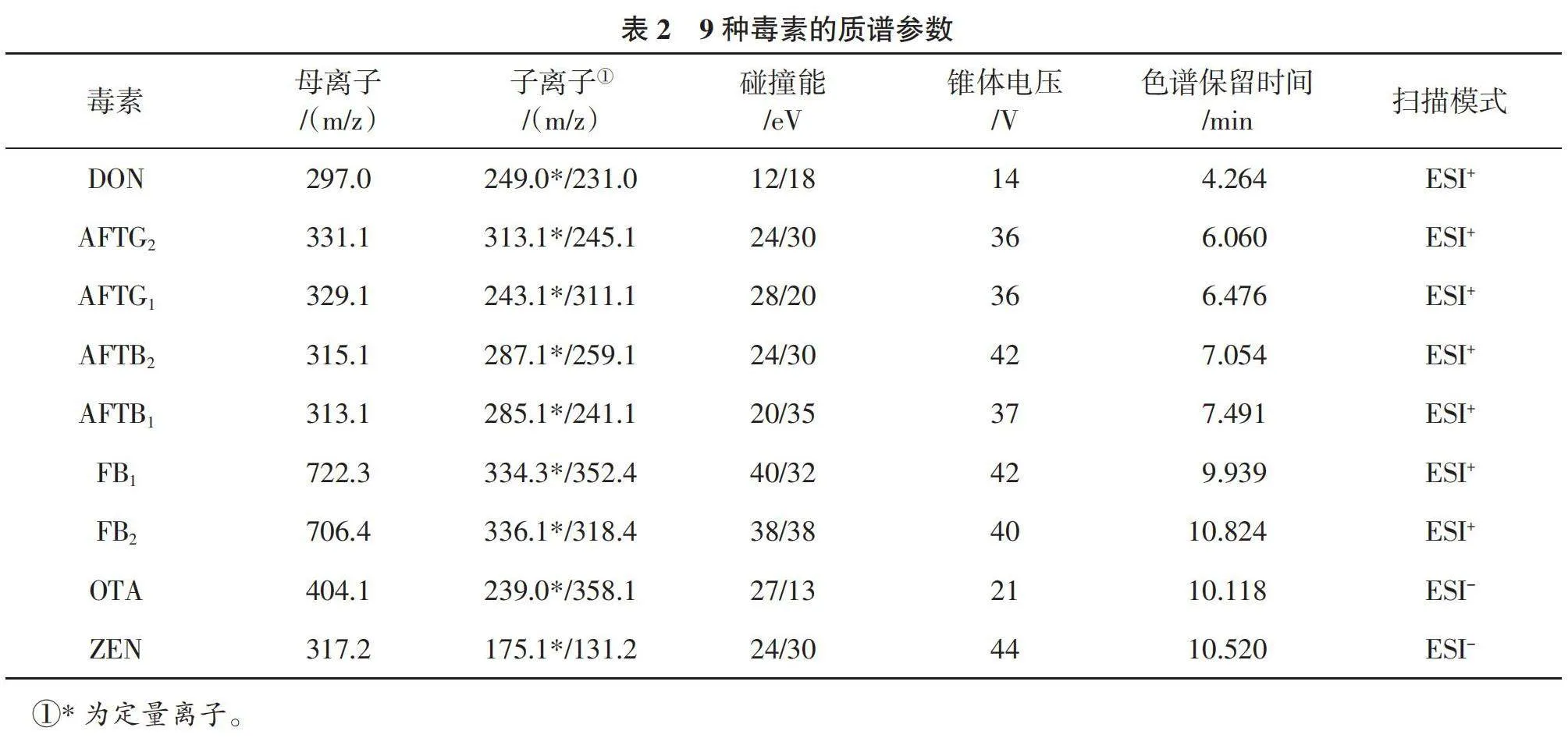
1.2.4 質譜條件 采用直接進樣技術,對9種目標化合物進行質譜條件的優化。分別在電噴霧離子化正模式(ESI+)和電噴霧離子化負模式(ESI-)下,對這9種真菌毒素的保留時間和峰形進行全面掃描。參考劉笑笑等[15 ]、李鳳華等[16 ]的方法,檢測方式為多反應監測(MRM);ESI掃描方式為正、負離子掃描。離子源溫度:正離子模式650 ℃、負離子模式600 ℃;氣簾氣:40 psi;電噴霧電壓:正離子模式4 000,負離子模式-4 000;柱流速0.3 mL/min,對照品溶液和當歸樣品溶液進樣量均為5 μl;9種真菌毒素質譜參數見表2。
1.2.5 流動相 參考2020版《中華人民共和國藥典》[17 ]、劉笑笑等[15 ]對比兩組流動相對9種真菌毒素出峰的影響情況。
1.2.6 凈化條件優化 分別使用微純生物科技有限公司和德國拜發公司的HLB固相凈化柱,對9種真菌毒素3個水平濃度的樣品進行凈化處理,每個濃度重復3次,比較9種毒素加標樣品的回收率。
1.2.7 方法學驗證 以1.2.2中混合對照品系列標準曲線濃度樣品,按照1.2.3和1.2.4測定法進樣測定,以平均峰面積為縱坐標,濃度為橫坐標,繪制標準曲線。據《中華人民共和國藥典》2020版四部通則9101分析方法驗證指導原則要求,把已知低濃度試樣測出的信號與空白樣品測出的信號進行比較,計算出能被可靠檢測出的被測物質最低濃度或量。以信噪比為3∶1時相應濃度或注入儀器的量確定檢測限;以信噪比為10∶1時相應濃度或注入儀器的量確定定量限。
1.2.8 方法精密度和回收率 分別取當歸空白基質溶液,添加定量限、2倍定量限和4倍定量限3個水平的9種真菌毒素混合標準溶液,進行前處理,測定其回收率和相對標準偏差(RSD),重復6次。
1.2.9 實際樣品的檢測 使用優化后的條件,對采集的72批當歸樣品9種真菌毒素同時進行檢測。
2 結果與分析
2.1 質譜條件
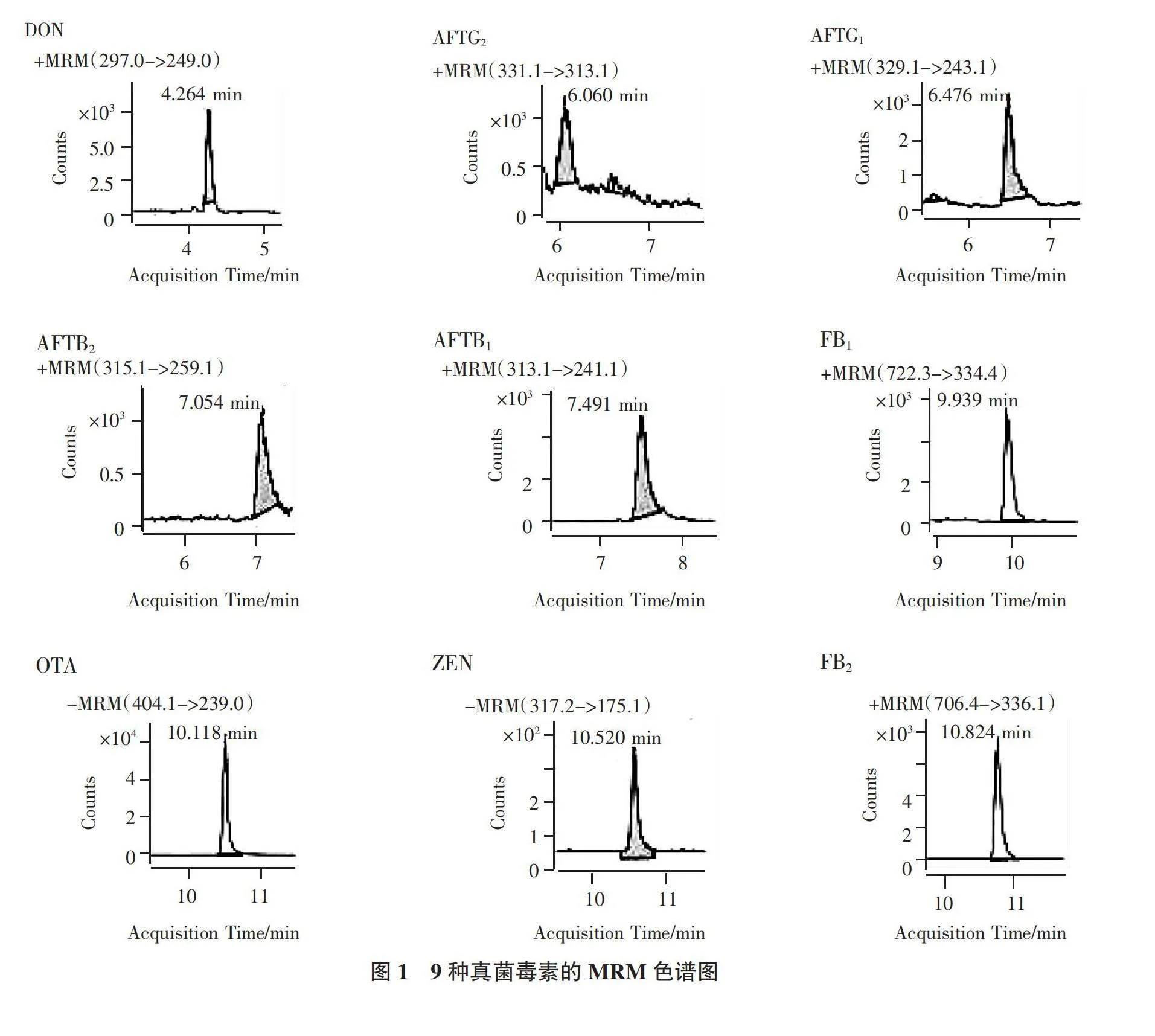
9種真菌毒素優化后的質譜參數見表2。優化時9種真菌毒素的濃度見表3,MRM色譜圖見圖1。可以看出,OTA、ZEN在ESI-掃描模式下響應更好,其余7種真菌毒素采用ESI+模式進行掃描。
2.2 流動相
由圖2可知,A組流動相(10 mmol/L醋酸銨溶液-甲醇)9種真菌毒素中FB2不出峰,其余成分均可正常出峰;B組流動相(0.1%甲酸的2 mmol/L甲酸銨-甲醇)9種真菌毒素均可正常出峰。因此,本方法選擇B組流動相。
2.3 凈化條件
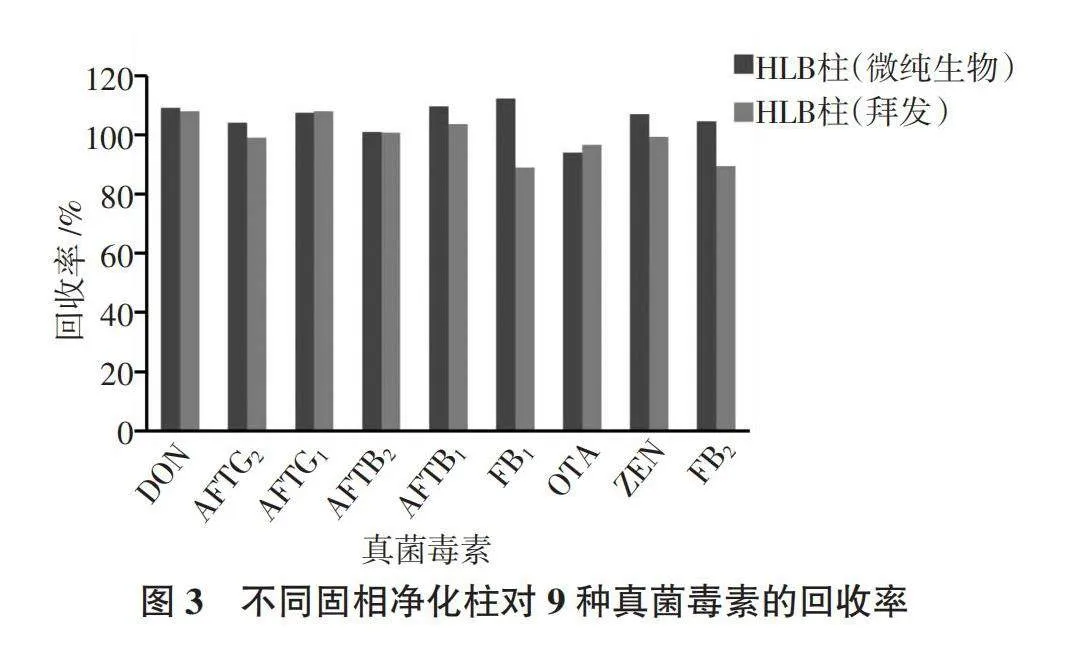
通過HLB柱(拜發)凈化的樣品較為澄清,通過HLB柱(微純生物)凈化的樣品略渾濁,說明HLB柱(拜發)的凈化效果略優于HLB柱(微純生物)。從圖3可以看出,FB1和FB2的檢測受基質效應影響,通過HLB柱(微純生物)凈化的樣品加標回收率略高于HLB柱(拜發),其余7個成分的兩組樣品加標回收率相近,符合2020版《中國藥典》第四部通則9101分析方法驗證指導原則要求,加樣回收率為70%~125%。兩種HLB柱都可以用于當歸9種毒素的測定,HLB柱(拜發)是毒素專用凈化柱,用于少量樣品的檢測;HLB柱(微純生物)成本相對較低,可用于大批量樣品的快速篩查和風險監測。
2.4 方法學驗證
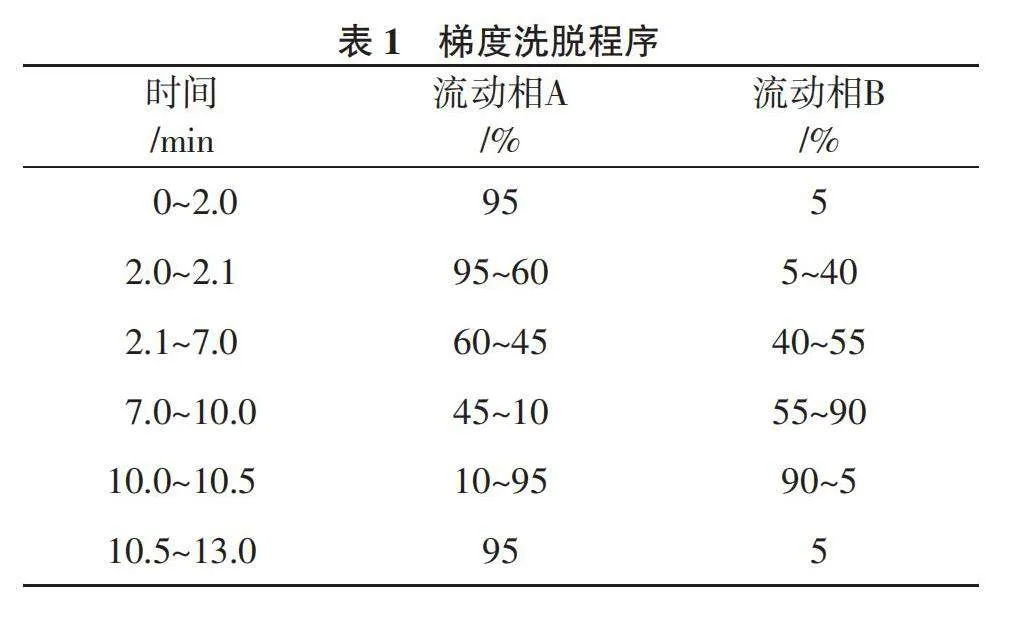
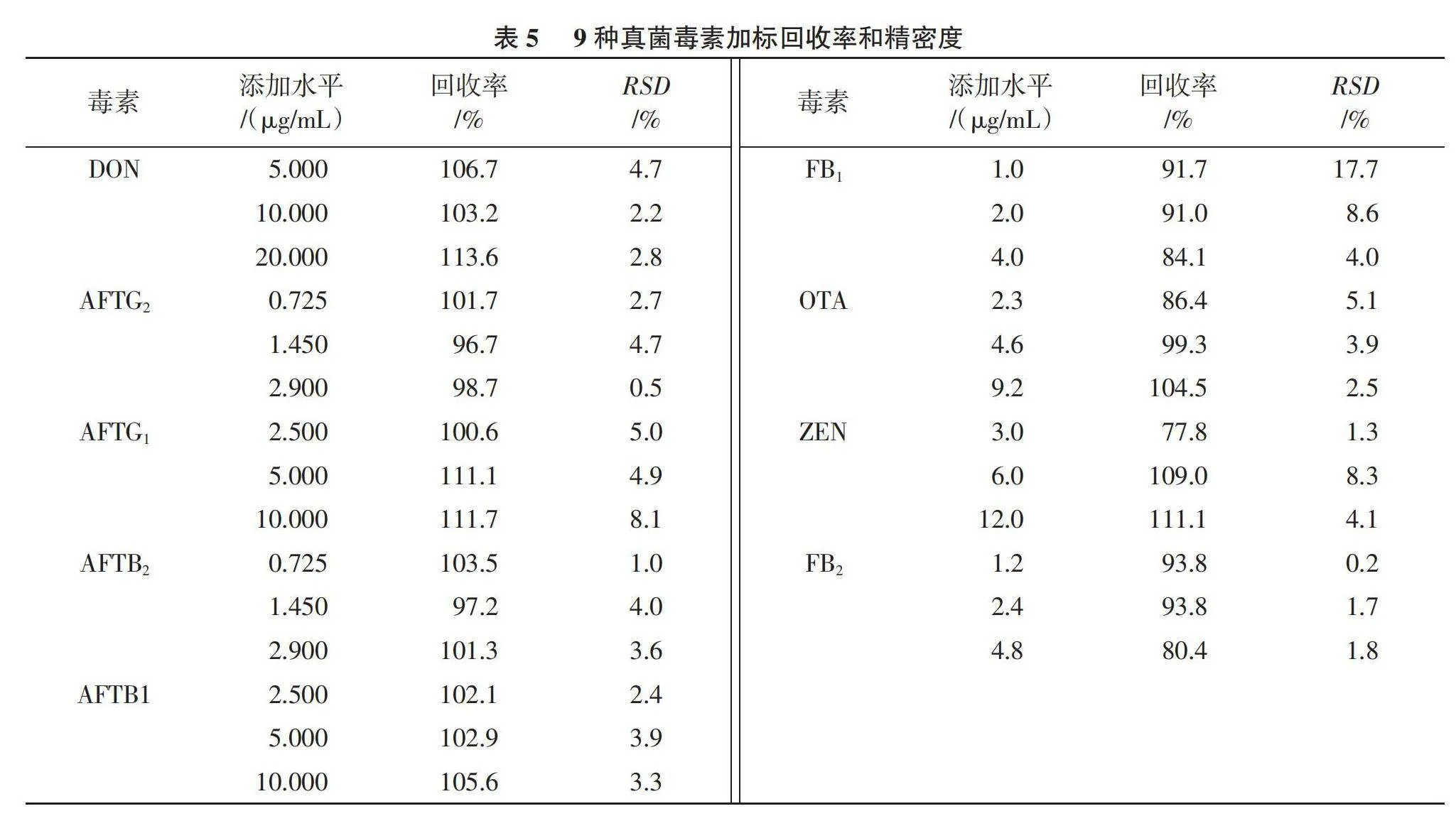
2.4.1 工作曲線和檢出限、定量限 由表4可以看出,在各自的線性范圍內,9種真菌毒素具有良好的線性關系,相關系數R2均大于0.995 0。方法的檢出限在0.04~1.43 μg/kg,定量限在0.12~4.75 μg/kg,均低于《中華人民共和國藥典》真菌毒素的限量標準[17 ],滿足限量檢出要求。
2.4.2 方法精密度和回收率 由表5可以看出,9種真菌毒素的加標回收率為77.8%~113.6%,測定值的相對標準偏差(RSD)為0.2%~17.7%,表明所建立的方法準確可靠。
2.5 樣品的檢測
通過對采集的72批當歸樣品檢測發現,有2批當歸樣品檢出真菌毒素,檢出率為2.8%,當歸中主要的污染毒素為AFTB1、AFTB2、AFTG1。2批次毒素檢出AFTB1濃度分別為3.3、3.4 μg/kg,AFTB2分別為7.2、7.1 μg/kg,AFTG1分別為10.4、9.6 μg/kg。2批次當歸AFT總量大于10 μg/kg,超出中藥材AFT總量≤10 μg/kg的限量。
3 討論與結論
在電噴霧質譜(ESI-MS)技術中,電離過程的溶液環境特性對分析結果的影響不容忽視[18 - 19 ]。首先,不同的溶劑系統可能會改變化合物在色譜柱上的吸附和解吸行為,從而影響其在色譜圖上的保留時間。其次,流動相的配比也會對峰形產生顯著影響。流動相中不同組分的比例變化可能會改變色譜柱的洗脫能力,進而影響目標化合物在色譜圖上的峰形。在電噴霧電離過程中,溶劑的極性和溶劑化能力對離子的形成和穩定至關重要。極性溶劑通常有助于形成更多的離子,而溶劑化能力的增強則有助于穩定這些離子,從而提高離子化效率。本研究通過樣品凈化條件,樣品前處理方法、色譜條件和質譜條件的優化,建立了一種利用固相萃取柱凈化、UPLC-MS/MS同時測定當歸中9種真菌毒素的方法。與《中華人民共和國藥典》和傳統方法中使用免疫親和柱相比,可較大程度降低檢測成本。樣品采用電噴霧電離源(ESI)和多反應監測模式(MRM),基質匹配外標法定量。9種真菌毒素標準曲線線性關系良好(R2 > 0.995 0),方法的檢出限為0.04~1.43 μg/kg,定量限為0.12~4.75 μg/kg,高、中、低3個濃度加標回收率為77.8%~113.6%,測定結果的相對標準偏差為0.2%~17.7%。該方法操作簡便,具有較好的凈化效果、良好的加標回收率、較高的準確度和可靠性,可滿足當歸中真菌毒素的大通量快速篩查和準確定量,也為其他中藥基質中多種真菌毒素含量的同時檢測提供技術借鑒。我們采用此方法對甘肅省72份不同來源的當歸中藥材樣本進行了系統檢測,通過檢測和數據分析,發現其中有2.8%的樣本存在真菌毒素污染,且主要污染的真菌毒素為黃曲霉毒素類(AFTB1、AFTB2、AFTG1)。這一結果表明,盡管大多數中藥材在采集和加工過程中能夠保持良好的衛生標準,但仍有部分樣本存在潛在的真菌毒素污染風險,本研究也為甘肅省當歸中真菌毒素污染狀況的調查提供了數據支持。
參考文獻:
[1] 高慧琴,張志紅,晉 玲. 十大隴藥(二)——當歸[J]. 甘肅中醫學院學報,2013,30(2):2.
[2] 楊董云,呂丙鈺,薛華麗,等. 新鮮當歸采后青霉病病原菌分離鑒定、生物學特性及毒素積累[J]. 甘肅農業大學學報,2022,57(2):74-81.
[3] 屠雄彪. 當歸不同藥用部位微量元素含量分析[J]. 中國民族民間醫藥,2011,20(3):43.
[4] 王雪梅,李應東. 當歸有效成分及其藥理作用的研究進展[J]. 甘肅中醫,2009,22(11):50-51.
[5] 陳書珍,季緒霞,楊成德,等. 甘肅省岷縣當歸病害調查及葉斑病田間藥劑篩選[J]. 草業科學,2017,34(12):2470-2475.
[6] 劉震營,張永清. 中藥材黃曲霉毒素污染與防控[J]. 山東中醫藥大學學報,2021,45(4):547-553.
[7] 吳潤松,葉 聰,胡綺萍,等. 中藥材真菌毒素污染檢測研究進展[J]. 廣東化工,2024,51(4):76-79.
[8] 劉麗娜,李海亮,李耀磊,等. 中藥真菌毒素質量控制概況、限量標準制定及有關問題的思考[J]. 中草藥,2023,54(19):6197-6207.
[9] 王文珺,孫雙艷,葉 金,等. 我國現行真菌毒素檢測標準概述[J]. 食品安全質量檢測學報,2019,10(4):837-847.
[10] 劉 靜,朱葉梅,董勝強,等. 液相色譜-串聯質譜法測定中藥材中11種真菌毒素[J]. 化學分析計量,2022,31(10):15-20.
[11] 黃仁堂,劉洪美,關凱儀,等. 液相色譜-串聯質譜法同時檢測中藥中多種真菌毒素的研究進展[J]. 中華中醫藥刊,2022,40(12):27-35;281.
[12] 范妙璇,董嬌嬌,王京輝,等. QuEChERS-超高效液相-三重四極桿串聯質譜測定白茅根中16種真菌毒素[J]. 中國中藥雜志,2017,42(19):3770-3775.
[13] 毛 丹,葉林鏈,王少敏,等. LC-MS/MS法同時測定中藥肉豆蔻中12種真菌毒素[J]. 中國藥師,2020,23(7):1311-1315.
[14] 胡佳哲,吳鳳丹,陳 俏,等. 同位素標記-高效液相色譜-串聯質譜法測定中藥材中8種真菌毒素[J]. 中國衛生檢驗雜志,2020,30(5):513-517.
[15] 劉笑笑,丁 輝,吳福祥,等. 雜質吸附固相萃取-液相色譜串聯質譜法同時測定糧食中15種真菌毒素[J]. 糧油食品科技,2021,29(1):155-167.
[16] 李鳳華,李作華,楊 麗,等. 藥食同源中藥材中16種真菌毒素的測定與分析[J]. 食品工業科技,2022,43(9):268-275.
[17] 國家藥典委員會. 中華人民共和國藥典:四部[M]. 北京:中國醫藥科技出版社,2020.
[18] 林 華,何 暢,吳易君,等. 超高效液相色譜-串聯質譜法測定紅曲中米酵菌酸[J]. 化學分析計量,2024,33(9):38-42.
[19] 冼燕萍,陳立偉,羅東輝,等. UPLC-MS/MS測定腐竹和米粉中的烏洛托品[J]. 江南大學學報(自然科學版),2012,11(1):78-82.
