磷酸化蛋白質的定量與化學計量分析方法的研究進展
2024-09-02 00:00:00劉媛翟睿吳帆楚占營趙洋戴新華方向俞曉平
分析化學 2024年5期
摘要 蛋白質翻譯后修飾是賦予蛋白質生理功能的關鍵機制,其中可逆的磷酸化修飾在眾多生命活動中起著開/關的作用,其異常變化通常與多種重大疾病過程密切相關。近年來,借助蛋白質組學技術和方法,磷酸化蛋白質的高通量、高精度定性與定量分析方法也得到了快速發展。本文綜述了近年來基于“自下而上”策略的磷酸化蛋白質定量與化學計量分析方法的研究進展,包括磷酸化肽的富集方法、磷酸化肽質譜碎裂方式、定量分析方法以及磷酸化位點的化學計量分析方法,并對磷酸化蛋白質研究的發展趨勢進行了討論。
關鍵詞 磷酸化蛋白質;富集方法;質譜;定量方法;化學計量;評述
蛋白質翻譯后修飾(Post-translational modifications, PTMs)是賦予蛋白質生理功能的關鍵機制,包括磷酸化、糖基化和泛素化乙酰化等[1]。PTMs 在信號轉導、細胞周期、發育與分化、細胞凋亡以及多種代謝通路調控中發揮了極為重要的作用[2-3]。經磷酸化修飾的蛋白質在催化、調節及蛋白相互作用等方面發揮著重要作用,是很多生命過程的關鍵調控“開關”[4],許多重大疾病(如心血管疾病、癌癥、神經退行性疾病等)已被證明與一些蛋白質的磷酸化水平變化密切相關。因此,對磷酸化蛋白質的定量與化學計量分析對理解生命體代謝過程、疾病發生機制以及個體預防治療等均具有重要意義[5]。
質譜技術具有高靈敏度、高精密度和高通量等優勢,已被廣泛用于磷酸化蛋白質的定量分析。然而,由于磷酸化蛋白質的豐度極低,并且易受到生物基質中高豐度肽段信號的干擾,采用質譜技術很難對磷酸化肽直接進行檢測[6]。因此,對磷酸化肽進行高效、高特異性富集是實現對其準確定性和定量分析的必要條件。針對上述問題,研究者開發了多種針對磷酸化肽富集和定量分析的方法,以提高磷酸化肽的質譜檢測靈敏度和定量分析的準確性,在此基礎上,開發了不同的質譜分析技術和基于“自下而上”
策略的磷酸化蛋白質定量和化學計量分析方法。本文介紹了磷酸化肽常用的富集方法、碎裂方式以及定量方法,總結了磷酸化位點的化學計量學及其研究方法近年來的研究進展,并討論了其未來的發展趨勢。
1 磷酸化肽的分離富集方法
目前,對于磷酸化蛋白質的定量和化學計量分析多采用“自下而上”的研究策略,首先將蛋白質酶解成肽段,再利用質譜技術對這些肽段進行分析,實現磷酸化蛋白質的鑒定和研究。對磷酸化肽段進行高效、高特異性的富集分離是實現高精度定性和定量分析的關鍵步驟[7]。研究者已基于磷酸化肽段化學特性開發了不同富集方法,大體可分為金屬離子親和色譜法、離子交換色譜法和免疫親和富集法(作用于pTyr 結構域)等[8]。
固定化金屬親和色譜法(Immobilized metal-ion affinity chromatography, IMAC)[9]和金屬氧化物親和色譜法(Metal oxide affinity chromatography, MOAC)[10]是兩種最普遍的富集pSer、pThr 和pTyr 的親和色譜技術[11]。IMAC 方法利用螯合作用將金屬陽離子(Fe3+、Ga3+和Zr4+等)固定在磁珠、二氧化硅或樹脂等基質上,再通過與帶負電荷磷酸基團的親和作用實現對磷酸化肽的選擇性保留。MOAC 主要是利用兩性金屬氧化物(如TiO2、ZrO2 和Fe3O4 等)[12]和磷酸根基團的結合作用富集磷酸化肽。Jensen 等[13]發現IMAC 更適宜多磷酸化肽富集而MOAC 更適宜單磷酸化肽富集。Thingholm 等[14]基于這兩個特點,將這兩種技術聯用,有效增加了磷酸化肽鑒定覆蓋率。
離子交換色譜技術是基于帶負電荷的磷酸化肽與色譜基質上的離子交換基團發生靜電相互作用,主要有強陽離子交換法(Strong cation exchange, SCX)和強陰離子交換法(Strong anion exchange, SAX)。Quan 等[15]發現SCX 主要是吸附一些堿性磷酸化肽段(pIgt;8.0),而SAX 主要是吸附一些酸性磷酸化肽段(pI 介于2.91~6.45 之間)和一些強酸性磷酸化肽段(pIlt;3.0)。免疫親和富集方法基于抗原-抗體反應,其特異性更強,但由于抗體開發周期長、價格昂貴以及結果重現性不佳等問題,主要用于富集豐度較低的酪氨酸磷酸化肽(pTyr)。SH2 結構域是已知最大的一類pTyr 識別結構域[16]。Bian 等[17]使用SH2 超親體并結合Ti4+-IMAC,實現了pTyr 的高特異性富集,從9 個人類細胞系中鑒定了約20000 個不同的pTyr和1000 余個酪氨酸磷酸化位點,其中36%的位點為首次鑒定。
N-磷酸化發生在堿性氨基酸上,形成磷酰胺鍵(P—N)。針對N-磷酸化肽的富集,已開發出pHis 抗體[18]和pArg 抗體[19]用于實現pHis 和pArg 肽段的有效富集。此外,基于IMAC 材料親和色譜保留時間差異的方法也被用于pHis 和pLys 肽段的有效富集[20-21]。為了提高N-磷酸化肽的覆蓋率,研究者又開發了多種富集N-磷酸化肽的方法。如Hu[22]等制備了具有核殼結構的亞二微米硅球,在硅球表面鍵合雙二甲基吡啶胺雙鋅分子,在中性條件下基于該材料的On-tip 富集方法實現了N-磷酸化肽段的高效、高選擇性和快速富集,結合液相色譜-質譜聯用分析技術,從HeLa 細胞中鑒定到3384 個N-磷酸化位點。然而, P—N 磷酰胺鍵具有較高的吉布斯自由能,在富集過程中易發生水解,因此,對于N-磷酸化肽的高效富集和檢測仍是一項具有挑戰性的研究工作。
2 磷酸化肽的質譜碎裂方式
磷酸化肽離子的二級碎裂解離和解析是“自下而上”的磷酸化蛋白鑒定與定量分析策略的關鍵,因此磷酸化肽離子的碎裂方式在分析檢測磷酸化位點過程中至關重要[23]。常用的磷酸化肽離子質譜碎裂方式包括碰撞誘導解離(Collision-induced dissociation, CID)、高能碰撞誘導解離(Higher-energy collisionaldissociation, HCD)、電子捕獲解離(Electron capture dissociation, ECD)、電子轉移解離(Electrontransfer dissociation, ETD)和紫外光誘導光解離(Ultraviolet photodissociation, UVPD)等。
CID 和HCD 是質譜實驗中常用的磷酸化肽碎裂方法,均通過碰撞裂解肽段生成碎片離子,以獲得關于肽段的結構和序列信息。CID 是通過氣相肽離子與惰性氣體(例如氦、氮)碰撞增加肽離子的內能,進而使肽離子碎裂[24-25]。雖然CID 的效率高,易于實現,但是CID 碎裂會造成磷酸化肽母離子及其b 型和y 型離子的非序列性中性丟失,丟失的中性部分可能是磷酸基團,影響磷酸化狀態和結構的分析。HCD 是通過高能碰撞誘導解離產生碎片離子,常用于離子阱式質譜儀(如Orbitrap),利用多極碰撞單元或碰撞池實現, HCD 碰撞的能量比CID 強很多,因此會產生更多的碎片離子[26],與CID 相比, HCD 碎裂中b 型和y 型離子仍然會經歷中性丟失,盡管中性丟失發生的頻率或程度較小,但仍會阻礙鑒定和位點定位[27]。
ECD 和ETD 都是利用電子與磷酸化肽離子的相互作用實現碎裂。ECD 依賴于多電荷氣相陽離子對低能電子的捕獲,通過低能量的自由電子與質子化的多電荷蛋白質或肽離子相互作用放熱而瞬間發生碎裂,主要產生由N—C 鍵斷裂形成的c 和z 類型碎片離子[28]。Kruger 等[29]發現CID 能產生更多的碎片離子,而ECD 能解離不同類型的化學鍵,將ECD 與CID 聯用可提高磷酸化蛋白測序的效率與準確度,增加磷酸化肽段鑒定覆蓋率。ETD 利用低電子親和力的陰離子將電子轉移到多電荷肽陽離子,使其發生裂解,通過離子阱可有效地捕獲陰離子試劑和陽離子肽[30]。主要產生由N—C 鍵α 斷裂形成的c 和z 類型碎片離子,可以有效保留肽段骨架信息和磷酸化修飾信息,同時克服了ECD 中熱電子傳遞和轉移時間長的缺點。ECD 和ETD 在碎裂磷酸化肽中主要存在兩個問題:(1)ECD 和ETD 的碎裂效率均取決于母離子的電荷狀態,對于三電荷及更高電荷的肽都表現出良好碎裂效率,但對于最常見的雙電荷磷酸化肽的碎裂效率不足;(2)ECD 和ETD 碎裂頻率較低,需要較長的反應時間才能實現母離子解離,相對于碰撞碎裂的方法需要更長的循環時間,對鑒定磷酸化肽的數量有影響[31]。
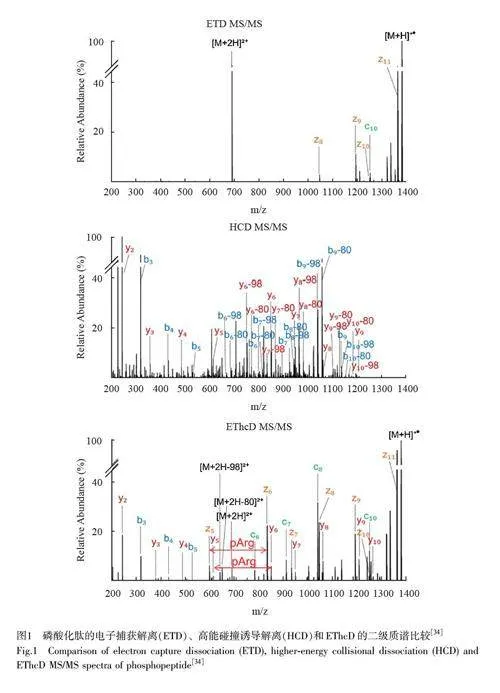
EThcD 融合了ETD 和高能碰撞解離HCD 兩種技術,其原理是先將較低能量的電子轉移解離,保留更多的修飾位點信息,然后通過高能碰撞解離產生更多的碎片離子,提供更詳細的序列信息,目前被廣泛應用于磷酸化蛋白質組學[32-33]。雖然EThcD 鑒定的磷酸化肽數目少于HCD,但其序列覆蓋率和位點定位的置信度比ETD 和HCD 顯著增加。磷酸化肽段經ETD、HCD 和EThcD 碎裂后的MS/MS 譜圖見圖1,經ETD 碎裂后磷酸化肽段序列覆蓋率較低,經HCD 碎裂產生足夠多的b 離子和y 離子,但伴隨著大量中性損失;經EThcD碎裂后的光譜圖提供了足夠的序列覆蓋,且沒有顯著的修飾中性損失[34]。Penkert等[34]使用含有磷酸化的合成肽驗證了EThcD 技術碎裂雙電荷磷酸化肽的能力,證明EThcD 在雙電荷磷酸肽的碎裂過程中優于ETD,并認為EThcD 是鑒定蛋白質中不穩定的磷酸化新位點的首選方法。
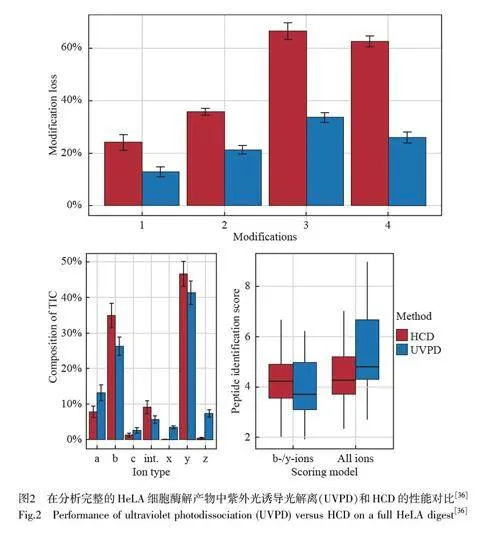
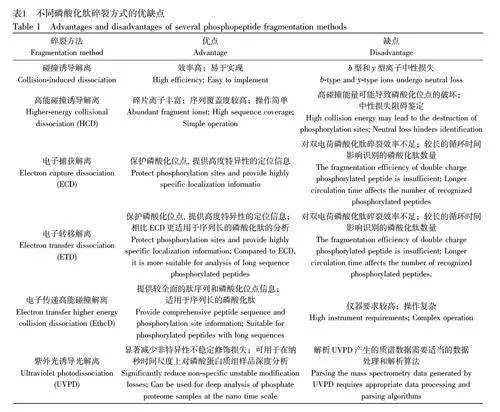
UVPD 是利用紫外光激發磷酸化肽內的鍵,引發磷酸化肽的離子解離,產生碎片離子,通過質譜對離子質荷比進行分析得到磷酸化位點和序列信息[35]。與HCD 相比, UVPD 產生的修飾損失顯著減少(如單、雙、三和四磷酸化肽離子分別減少47%、40%、50%和58%), b 離子和y 離子損失也較少,并且顯示的離子得分顯著增加,如圖2 所示[36]。相比HCD, UVPD 提供的譜圖信息更多,能顯著減少磷酸化修飾基團的損失,并實現在納秒時間尺度內深度分析磷酸化蛋白質組樣品[36]。Robinson 等[37]發現,與HCD 相比, UVPD 提供了更多類型的子離子,并改善了子離子上的磷酸基團信息的保留,雖然陰性模式UVPD 在檢測和測序高酸性磷酸化肽方面更優,但由于陰性模式下的電離效率較低,在進行大規模分析時可能無法充分覆蓋整個樣品的范圍,因此, UVPD 適于與其它質譜技術結合(如HCD),充分利用各自的優勢,提高磷酸化蛋白質的檢測和測序能力,從而獲得更可靠的結果。不同磷酸化肽碎裂方式的優缺點見表1。
3 磷酸化蛋白質的定量分析方法
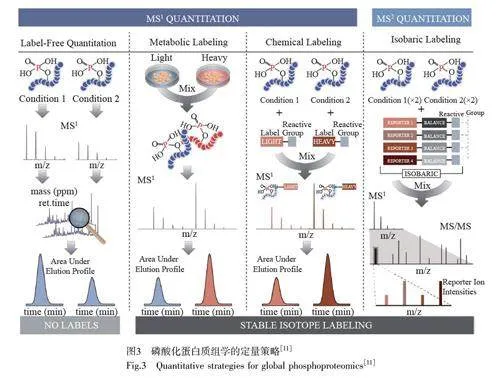
磷酸化蛋白質的定量分析主要采用“自下而上”的研究策略。相較于蛋白質組,磷酸化蛋白質組的定量分析更困難,具體表現在:(1)生物樣品中非磷酸化肽含量較多,干擾磷酸化肽的質譜信號;(2)由于磷酸化肽段具有負電性,在質譜正離子模式下較難帶正電荷,離子化效率低;(3)磷酰鍵比肽鍵更易斷裂,增加了磷酸化肽定量的難度[38]。磷酸化肽段的定量分析方法有很多種,根據所用的質譜信息可分為一級質譜定量(MS1)和二級質譜定量(MS2),如圖3 所示[11]。根據前處理過程的不同, MS1 定量又可分為非標記定量(Label-free)、代謝標記定量(Metabolic labeling)和化學標記定量(Chemical labeling)等;MS2定量主要是等重標記定量法。
3.1 非標記定量方法
Label-free 方法的準確性主要取決于液相色譜-質譜聯用系統的穩定性。Label-free 主要有兩種方法:其一為譜圖計數法,對獲得的目標肽段二級質譜圖數量進行計數和比較,以此作為蛋白質表達量差異的定量依據;其二為離子強度法,利用測定的多肽母離子的色譜峰面積實現對蛋白質的表達量的定量分析[39]。與標記法相比, Label-free 無需使用特殊介質或昂貴的試劑,避免了在標記過程帶來的實驗誤差,同時不需要考慮標記效率[40]。但是, Label-free 數據處理過程復雜、對儀器性能要求高、不適用于低豐度蛋白質檢測。目前,已有研究者將Label-free 應用于重大疾病相關磷酸化蛋白質的定量分析,如Liu 等[41]利用高分辨率質譜法和開放式搜索方法對胃癌患者的腫瘤及癌組織的磷酸化蛋白質進行Label-free 定量分析,共鑒定了832 個與胃癌相關的磷酸化位點,為尋找放射治療耐藥性的重要靶點奠定了基礎。此外, Label-free 也應用在識別生物標志物方面,如De Oliveira 等[42]為識別胃腺癌患者血清生物標志物,使用Label-free 對胃腺癌患者血清和非癌癥患者血清中的33 種肽進行了差異定量分析,其中,19 種肽在胃腺癌患者血清樣品中表達上調, 14 種肽表達下調。該研究表明,蛋白酶MMP-7 是一種有前景的生物標志物。
3.2 代謝標記定量方法
代謝標記定量方法是通過細胞正常代謝使蛋白質帶上同位素標簽。典型的代謝標記定量方法為氨基酸穩定同位素標記法(Stable isotope labeling by amino acid in cell culture, SILAC)[43]。SILAC 將輕、中或重型穩定同位素標記的必需氨基酸(賴氨酸和精氨酸)加入細胞培養基,通過細胞的正常代謝,新合成的蛋白會帶上穩定同位素標簽,將各類型蛋白質等量混合后進行質譜分析,通過比較一級質譜圖中同位素峰形的面積而進行相對定量分析[44]。SILAC 的主要優勢是在實驗樣品前處理時進行標記,從而將平行樣品處理過程中可能引起的誤差降至最低。但是, SILAC 需要經過多次細胞分裂才能確保定量分析穩定,因此不適合定量分析初代細胞。由于SILAC 可用于分析細胞、組織和體液等,因而被廣泛應用于表征不同的生物樣本蛋白質磷酸化差異[45]。Stepath 等[46]評估了Label-free、SILAC 和TMT 方法對磷酸化位點量化方面的性能,發現SILAC 在磷酸化位點的量化方面表現出較高的精度,是分析細胞培養模型中細胞信號傳導的首選方法。Erber 等[47]利用SILAC 技術研究海馬神經元HT-22 細胞中急性和慢性缺鐵引起的磷酸化信號通路的變化,發現超過10%的磷酸化位點的相對豐度變化超過2 倍,表明缺鐵和缺氧顯著影響神經元細胞中的磷酸化信號通路。一些SILAC 衍生方法也被應用于磷酸化蛋白質組學分析,如脈沖式穩定同位素標記技術(Pulsed SILAC, pSILAC)[48]、高級穩定同位素標記技術(Super-SILAC)[49]等。與SILAC 直接分析混合后的輕重標記的樣品不同, Pulsed SILAC 技術是在細胞培養過程中將“輕”、“中”、“重”氨基酸交替加入到培養基中,并培養特定時間,以跟蹤和定量分析蛋白質的動態變化[50]。Wu 等[51]基于Pulsed SILAC 開發出差異同位素標記技術(DeltaSILAC)新策略,將細胞在不同時間點進行SILAC 標記,比較不同時間點的蛋白質組成的變化,該方法可以直接考察磷酸位點對蛋白質壽命的影響,進一步分析磷酸化位點對蛋白質表達壽命的影響。
3.3 化學標記定量方法
化學標記定量方法是采用物理和化學性質相似的化學試劑對生物樣品進行標記,再采用液相色譜-質譜聯用系統對生物樣品進行定量分析。近十年來,化學同位素標記受到越來越多的關注,并實現了對生物樣品中內源性代謝物的準確定量分析[52]。磷酸化蛋白質組學中最常用的化學標記定量方法是穩定同位素二甲基標記定量分析方法(Stable-isotope dimethyl labeling),該方法具有良好的標記效率、簡單的實驗過程以及低廉的試劑成本,可用于分析多種類型樣本。二甲基標記采用甲醛(CH2O)和氰代硼氫化鈉(NaBH3CN)兩種穩定的同位素試劑標記蛋白質/肽的N 端或賴氨酸的氨基基團,使蛋白質/肽標記上不同的標簽[53],可在每個標記位點與非標記對應物間產生28 個質量單位值差,在每個對應標記同位素上產生4 個質量單位值差,并且不會產生任何可檢測的副產物[54]。由于二甲基適用性強,因而被廣泛應用于重大疾病的磷酸化組學定量分析。Liu 等[55]使用穩定同位素二甲基標記結合高分辨率質譜法,在5-Fu耐藥細胞系Bel/5-Fu 中共確定了8272 種獨特的蛋白質和22095 個具有高定位置信性的磷酸化位點,并確定了肝細胞癌中減弱化學耐藥性的潛在靶點。
3.4 等重標記定量方法
等重標記定量方法的原理是基于報告區與平衡區具有相同數量的總重同位素,當報告離子斷開后即可通過比較樣品之間相對報告離子強度對樣品進行定量分析,包括同位素標記相對和絕對定量分析(Isobaric Tags for relative and absolute quantitation, iTRAQ)[56]、串聯質譜標簽(Tandem mass tags, TMT)[57]、氘同位素標記定量分析(Deuterium (2H) isobaric aminereactive tag, DiART)以及組合等壓質量標簽(Combinatorial isobaric mass tags, CMTs)等。
3.4.1 同位素標記相對和絕對定量分析
同位素標記相對和絕對定量分析(iTRAQ)的原理是利用多種同位素試劑標記蛋白多肽N 末端或賴氨酸側鏈基團,經高精度串聯質譜分析實現蛋白質組定量分析。目前, iTRAQ 試劑有4-plex 和8-plex,可分別標記4 組或8 組樣品,包含與氨基結合的反應基團、不同分子量的報告基團以及不同分子量的質量平衡基團,當不同的報告基團分別與對應的平衡基團結合后會達到相同的質量,但在MS/MS 碎裂后產生獨特的報告離子,以此對多肽進行定量分析[58]。iTRAQ 標記無偏倚且掃描范圍廣,可用于絲氨酸、蘇氨酸和酪氨酸位點上的磷酸化定量分析[59]。為了鑒定參與調節MAT-LyLu 細胞轉移的關鍵生物標志物,Xu 等[60]分別用TTX-S VGSCs 的特異性抑制劑HNTX-Ⅲ和激活劑JZTX-Ⅰ處理細胞,然后進行了基于iTRAQ 的定量磷酸化蛋白質組學分析,與對照組相比,在HNTX-Ⅲ和JZTX-Ⅰ處理組中分別鑒定出554 種和1779 種獨特的磷酸化蛋白,其中, 55 種和36 種磷酸化蛋白質被鑒定為差異表達的蛋白質。差異表達的磷酸化蛋白與前列腺腫瘤的遷移和侵襲顯著相關,因此有望成為前列腺癌癥精確藥物的潛在生物標記物。
3.4.2 串聯質譜標簽
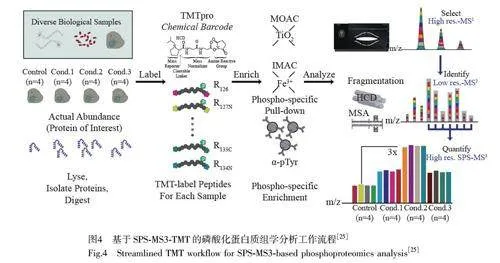
TMT 標記具有通量高、準確性高、對低豐度蛋白的鑒定效率高等優點,可同時對多個樣品進行全蛋白質組定量分析,最新的16-Plex TMT 標記允許對多達16 個樣本進行蛋白質組定量分析[61]。基于SPS-MS3-TMT(Synchronous precursor selection-MS3)的磷酸化蛋白質組學分析的主要流程如圖4 所示[25]。TMT 標記定量磷酸化肽的方法已被廣泛應用。Friedrich 等[62]使用TMT 標記,在非小細胞肺癌、亞型腺癌和鱗狀細胞癌中定量分析8000 余種蛋白質,并確定了超過1400 個磷酸位點,為肺癌相關癌基因、腫瘤抑制因子及信號通路的蛋白質豐度和磷酸位點調節提供了相關定量信息。人參皂苷是一種重要的抗癌活性成分, Zou 等[63]利用TMT 標記定量分析人參皂苷治療后乳腺癌MDA-MB-231 細胞中磷酸化蛋白質組的變化,在皂苷處理后的MDA-MB-231 細胞中鑒定出13 個位點的磷酸化狀態發生變化,分析了人參皂苷的抗癌作用機制。TMT 標記反應通常在磷酸化肽富集之前,但TMT 標記對肽標記后會改變其化學性質,從而導致TiO2 富集磷酸肽的選擇性發生變化, Ogata 等[64]設計了一種納米級固相TMT 標記反應器,在磷酸化肽富集后進行TMT 標記,通過優化固相TMT 標記反應器中的pH 值、反相吸附劑和離子成功標記磷酸化肽,只需要少量TMT 試劑標記磷酸肽即可完成測定。此研究有助于納米級TMT 標記應用于大規模磷酸蛋白質組學研究。
3.5 微量樣品中磷酸化蛋白質的定量策略
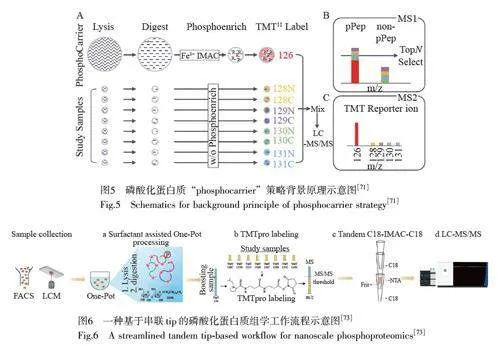
磷酸化蛋白質定量組學分析通常采用數據依賴采集模式(Data dependent acquisition, DDA)采集質譜數據,在該模式下,需要有足夠高豐度的磷酸肽離子才能實現有效的MS1 和MS2 分析。由于生物樣本中磷酸化蛋白質豐度低,并且在正離子模式下質譜對磷酸化肽離子化效率較差,目前多需使用300~1000 μg 的提取蛋白才能實現可重復性的深度磷蛋白質組鑒定[65]。然而,臨床樣品通常只能提供少于10 μg 的提取肽,在如此微量的樣品中實現磷酸化蛋白定量分析是一項重要且艱巨的任務。Budnik 等[66]利用等重標記法的MS1 信號強度取決于所有通道混合后的肽段總含量且各樣品的MS2 定量離子信號互不干擾這一特性,開發了一種高通量SCoPE-MS(Single cell protEomics by mass spectrometry)方法,用于分析單細胞蛋白質組。選擇TMT 標簽的其中一個通道標記200 個細胞樣品作為“Carrier”以提高MS1 信號強度,其余通道標記待測單細胞樣品,所有通道混合后進行定量分析,在單細胞水平的蛋白質組學分析實驗中,實現了對平均細胞直徑僅為11 μm 的Jurkat 和U-937 細胞的蛋白質定量分析。上述的“Carrier”策略為微量樣本中磷酸化蛋白質組的定量分析提供了新思路[67-69]。Yi 等[70]開發了一種BASIL(Boosting to amplify signal with isobaric labeling)等重標記策略,選擇TMT 的一個通道標記“Carrier”樣品,與其它通道混合后進行磷酸化肽富集和質譜檢測,該設計顯著增強了磷酸化肽的可檢測性和可識別性,使可量化磷酸化位點的總數增加了4 倍以上。但是,在BASIL 策略中,研究樣品和“Carrier”樣品是在混合之后進行磷酸化肽富集和檢測,每次分析都需要進行磷酸化肽富集處理,因而每個通道至少需要10 μg 的研究樣品才能滿足磷酸化肽分析的需求。Kwom 等[71]對上述策略進行了改進,使用富集后的高濃度磷酸化肽取代蛋白質組樣品作為“Phosphocarrier”樣品,該策略的原理示意圖如圖5 所示。來自“Phosphocarrier”樣品的高濃度磷酸化肽貢獻更強的MS1 信號強度(圖5B),研究樣品通道中的磷酸化肽無需進行富集處理且更易被選擇用于MS2 分析(圖5C),因而每個研究通道僅需低于1 μg 的樣品即可實現全面的磷酸化蛋白質組分析。
酪氨酸磷酸化僅占總O-磷酸化蛋白質的0.05%,因此微量樣品中酪氨酸的全面磷酸化蛋白質組學分析更困難。Chua 等[72]開發了BOOST(Broad-spectrum optimization of selective triggering)策略,利用酪氨酸磷酸酶抑制劑(Pervanadate, PV)處理的細胞作為TMT 標記的一個通道,選擇性地增加了含磷酸化酪氨酸肽段的信號相對豐度,使磷酸化酪氨酸的定量深度提高6.3 倍,并且從1 mg T 細胞受體激活的Jurkat T 細胞中定量分析了2300 多種特異性酪氨酸磷酸化肽。
前處理樣品轉移過程中產生的非特異性表面吸附導致的樣品損失也是微量樣品磷酸化蛋白質定量分析面臨的重要問題。Tsai 等[73]開發了一種基于串聯Tip 的磷酸化蛋白質組學樣品制備方法,該方法利用C18 與IMAC 結合形成串聯C18-IMAC-C18,可快速、高效地富集磷酸化肽,并與SOP(Surfactant-assistedone-pot)[74]方法和iBASIL(Improved Boosting to amplify signal with isobaric labeling)[75]方法結合形成了一個簡化的工作流程(圖6),實現了高靈敏度、高通量的納克級磷酸化蛋白質組定量分析,精確定量分析了100 個MCF10A 細胞中約600 條磷酸化肽和200 μm×200 μm×10 μm 的人類脾臟組織體素(相當于約100 個細胞)中約700 條磷酸化肽,為納克級磷酸化蛋白質組分析開辟了新途徑。
4 磷酸化位點的化學計量學
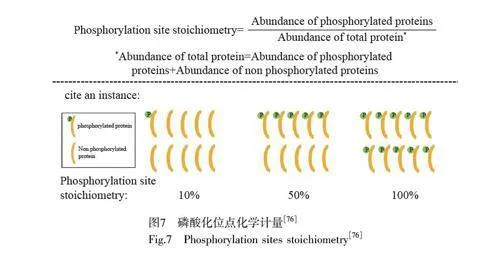
上述富集和定量方法幫助研究者鑒定到大量的磷酸化位點,并在一些研究中確定了磷酸化蛋白質的相對和絕對豐度。然而,蛋白的功能除了與特定位點的化學修飾有關外,還與該位點的修飾程度有關[76]。磷酸化位點占有率也稱磷酸化位點化學計量,可通過測量給定位點的磷酸化形式相對于蛋白質總量的比例實現(圖7)[76]。磷酸化位點的化學計量是磷酸化的重要屬性,也是確定磷酸化位點生物學相關性的重要指標,對磷酸化依賴性的蛋白質調節過程研究具有重要意義[77]。與重大疾病相關的蛋白質磷酸化位點的化學計量研究對于深入理解疾病的發生機制以及開發新的治療方法具有重要意義[78-79]。
4.1 絕對定量分析
由于蛋白質組的復雜性,在蛋白質組水平上確定特定位點的絕對磷酸化化學計量是一項具有挑戰性的任務。早期,磷酸化化學計量均采用低通量方法,如定量蛋白質印跡法[80]。2003 年, Gerber 等[77]首次使用AQUA(Absolute quantification)法測量人體細胞周期依賴性的分離酶上Ser-1126 位點的磷酸化和非磷酸化形式的絕對量,從16 μg 樣品中定量人類分離酶蛋白Ser-1126 位點的化學計量為34%。該方法的原理是向待測樣品中加入已知量的穩定同位素標記磷酸化肽段和與其對應的穩定同位素標記非磷酸化肽段,通過比較同位素標記肽段與目標肽段的信號強度計算目標肽段的豐度。AQUA 被應用于精準量化不同樣本的磷酸化位點化學計量,也可用于新的磷酸化位點化學計量定量方法的評估。但是, AQUA 在量化磷酸化位點化學計量時會受到肽電離效率的影響,并且AQUA 定量肽合成的成本較高。
4.2 非標記定量分析
采用同位素標記肽段進行定量分析成本較高, Steen 等[81]開發了一種非標記定量的方法,用于估計樣品系列中磷酸化化學計量,當磷酸化肽及其對應的非磷酸化肽兩種肽的信號強度變化具有相關性時,可通過監測磷酸化肽及其對應的非磷酸化肽的離子電流強度來測量磷酸化化學計量,基于此設計了相關計算方案:首先采用適當的歸一化程序確定檢測效率,然后通過監測磷酸化肽及其對應的非磷酸化肽之間的信號強度比即可得到目標磷酸化肽段的化學計量。該方法可用于定量分析多重磷酸化肽或蛋白酶切割效率受磷酸化位點影響的肽。該方法的局限是:定量分析的前提條件是目標磷酸肽及其對應的非磷酸化肽兩種肽的信號強度變化必須相關;蛋白質的磷酸化比例必須達到一定程度(gt;10%),但不完全磷酸化。此外, Bekker-Jense 等[82]將多種實驗條件信息集成到一個化學計量模型中,通過使用多通路復用量化磷酸化肽、非磷酸化肽和相應的蛋白質信號強度,然后在Perseus 插件中實現線性建模和化學計量計算,最終使用戶能從Label-free 數據中計算磷酸化占用率。
4.3 磷酸化肽和非磷酸化肽的相對豐度分析
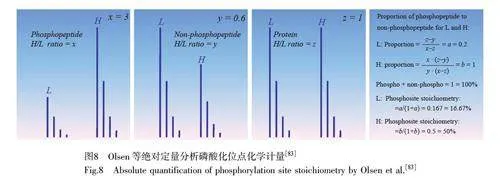
通過量化磷酸化肽和非磷酸化肽的相對豐度對磷酸化位點進行化學計量分析,通常是先量化樣品中的磷酸化肽、非磷酸化肽和相應的蛋白質強度,再結合比例關系進行數學計算,進而得到位點的磷酸化水平。Olsen 等[83]假設磷酸化肽段與其對應的非磷酸化肽段比例達到1,從SILAC 輕標和SILAC 重標兩種狀態下計算磷酸化位點的絕對化學計量,具體計算公式如圖8 所示,其中, x 表示SILAC 標記的輕、重磷酸化肽比例, y 表示SILAC 標記的輕、重非磷酸化肽比例, z 表示SILAC 標記的輕、重蛋白質比例,通過x、y、z 的SILAC 標記比率信息計算位點的磷酸化化學計量。采用該方法實現了磷酸化位點化學計量的大規模研究,并獲得了人類培養細胞系HeLa S3 細胞周期中蛋白質和磷酸化位點動力學的系統視圖。此外, Hogrebe 等[84]開發了一種基于三維多元回歸模型(3DMM)的方法,使用量化方法中的磷酸化肽、非磷酸化肽和相應的蛋白質強度,結合肽的數量與肽的強度比例關系,在不同條件下對這3 種強度進行多元回歸擬合,計算磷酸化程度。
近些年,已有大量通過磷酸酶去除磷酸化再結合同位素標記技術來測量蛋白質的磷酸化位點的研究[85-86]。Wu 等[87]將蛋白質裂解產物的兩個相同等分試樣分別進行虛擬處理以及磷酸酶處理,然后用穩定同位素進行差異化學標記。混合后,將獲得的肽段序列與已知位點的數據庫進行對比,基于重疊的非磷酸化形式的比率計算化學計量,最終確定了對數中期的釀酒酵母的5033 個磷酸化位點的化學計量。Chen 等[88]設計了一種二甲基標記結合磷酸酶去磷酸化的方法定量分析蛋白質中特定位點磷酸化水平,測得牛奶中α-S1 酪蛋白Ser130 位點和α-S2 酪蛋白Ser158 位點的磷酸化程度分別為97.2%和97.3%,這是首次在肽段水平上報道α-酪蛋白位點的特異性磷酸化程度。隨后,研究者采用該方法測得未經過氧化叔丁醇溶液(Tert-butyl hydroperoxide, t-BHP)處理的Hsp27 上Ser82 的磷酸化程度為10.76%,經t-BHP 處理的Hsp27 上Ser82 的磷酸化程度為37.46%,表明該方法在分析蛋白質磷酸化動態變化方面具有可行性。
上述磷酸酶處理和同位素標記的方法具有工作流程簡單、數據處理易于實現等優點,但僅適用于定量蛋白樣品上有限數量位點的化學計量。在通過量化修飾肽和未修飾肽的相對豐度來進行化學計量測定的方法中,確保觀察到的肽豐度變化具有統計學意義至關重要。為了使肽豐度變化更具有統計學意義, Lim 等[89]設計了貝葉斯模型(The Bayesian models),將化學計量建模為β 分布,可解決基于磷酸酶的磷酸化化學計量實驗中的負化學計量的問題。
4.4 同位素標記非磷酸化肽段
使用磷酸化標準肽段定量磷酸化位點的化學計量會面臨一些問題,其中合成定量用的磷酸化肽段中含有其對應的非磷酸化肽段,同時,磷酸化肽的低靈敏度也給質譜監測帶來挑戰。為規避上述問題,可通過磷酸酶處理或表達出無磷酸化修飾的QconCAT 蛋白,再通過歸一化計算提高定量分析的準確性,最后只需利用同位素標記的非磷酸化肽段即可對目標肽段進行化學計量定量分析[90]。除此之外,Dominik 等[91]開發了一種基于磷酸酶處理和同位素標記用于測定復雜生物樣品中的磷酸化化學計量的定量方法(Phosphatase-based phosphopeptide quantitation, PPQ)。該方法在酶切后加入同位素標記的非磷酸化肽段,然后將樣品分為未處理和磷酸酶處理兩部分,比較兩部分樣品的峰面積比即可確定磷酸化化學計量,因此,只需比較非磷酸化肽段的相對或絕對豐度即可確定單個肽的磷酸化化學計量。
4.5 磷酸化化學計量分析在激酶研究中的應用
激酶能夠轉移磷酸基團到特定的底物分子上,從而引發磷酸化修飾。磷酸化化學計量對于揭示激酶的調控機制和信號傳導網絡具有重要意義。激酶介導的磷酸化化學計量在細胞調控和疾病發展過程中具有重要作用。不同激酶對磷酸化位點的化學計量具有特異性,這種特異性可以調節底物的功能和細胞過程。此外,異常的磷酸化化學計量與許多疾病的發生發展密切相關,包括腫瘤、神經系統疾病和心血管疾病等。為了探究激酶對磷酸化的影響, Karayel 等[92]針對帕金森病患者中性粒細胞中Rab10-Thr73磷酸化水平進行了研究,發現帕金森病患者的富亮氨酸重復激酶2(Leucine-rich repeat kinase 2, LRRK2)突變會增加Rab10-Thr73 磷酸化水平。Tsai 等[93]采用兩次富集結合磷酸酶處理的方法探究了不同激酶作用位點的磷酸化化學計量,在使用CK2、MAPK 和EGFR 作為肺癌癥細胞中的基序靶向激酶的驗證實驗中,測量了1000 余個磷酸化位點,包括642 個CK2 靶向位點、940 個MAPK 靶向位點和366 個EGFR靶向位點,然后對不同激酶作用的磷酸化位點進行統計,發現CK2 有30%的磷酸化位點化學計量超過70%,而MAPK 和EGFR 只有少于15%的磷酸化位點化學計量大于70%,為激酶與磷酸化化學計量研究提供了新的研究思路。
5 總結和展望
磷酸化蛋白質參與多種生理和病理過程,具有重要的研究意義。不同類型的磷酸化肽段需采用不同的富集方法,質譜技術的發展也為磷酸化蛋白質研究提供了可靠的技術支撐。在此基礎上的磷酸化位點化學計量學研究不僅對探究磷酸化位點和占有率具有生物學意義,也可用于分析磷酸化位點上游激酶的靶向位點和作用機制,這些成果可應用于重大疾病發病機制的探索和活性激酶抑制劑的臨床研究,評估激酶抑制劑的靶向結合、劑量和療效等,在生物醫學上具有較大的應用潛力。此外,對微量樣品進行全面的磷酸化蛋白質組學分析仍然是一項艱巨的任務,也是目前的研究熱點,研究者采用不同的解決方案提高微量樣品檢測效率。磷酸化蛋白質的定量與化學計量分析方法在深入理解細胞信號傳導、生物學過程和疾病機制方面發揮關鍵作用,未來的研究方向應主要集中在提高分析方法的精確度、靈敏度和通量,開發治療重大疾病的靶向磷酸化位點的藥物以及加速其在生物醫學和精準醫學領域的應用。
References
[1] DUAN G Y, WALTHER D. PLoS Comput. Biol. , 2015, 11(2): 23.
[2] CZUBA L C, HILLGREN K M, SWAAN P W. Pharmacol. Ther. , 2018, 192: 88-99.
[3] SEYREK K, LAVRIK I N. Apoptosis, 2019, 24(5-6): 385-394.
[4] GRIMSRUD P A, SWANEY D L, WENGER C D, BEAUCHENE N A, COON J J. ACS Chem. Biol. , 2010, 5(1): 105-119.
[5] THORNER J, HUNTER T, CANTLEY L C, SEVER R. Cold Spring Harbor. Perspect. Biol. , 2014, 6(12): a022913.
[6] MANN M, ONG S E, GR?NBORG M, STEEN H, JENSEN O N, PANDEY A. Trends Biotechnol. , 2002, 20(6): 261-268.
[7] CHAN C Y, GRITSENKO M A, SMITH R D, QIAN W J. Expert Rev. Proteomics, 2016, 13: 421-433.
[8] LOW T Y, MOHTAR M A, LEE P Y, OMAR N, ZHOU H, YE M. Mass Spectrom. Rev. , 2021, 40(4): 309-333.
[9] FICARRO S B, MCCLELAND M L, STUKENBERG P T, BURKE D J, ROSS M M, SHABANOWITZ J, HUNT D F,WHITE F M. Nat. Biotechnol. , 2002, 20(3): 301-305.
[10] LARSEN M R, THINGHOLM T E, JENSEN O N, ROEPSTORFF P, J?RGENSEN T J. Mol. Cell. Proteomics, 2005, 4(7):873-886.
[11] RILEY N M, COON J J. Anal. Chem. , 2016, 88(1): 74-94.
[12] KANSHIN E, MICHNICK S W, THIBAULT P. J. Proteome Res. , 2013, 12(6): 2905-2913.
[13] JENSEN S S, LARSEN M R. Rapid Commun. Mass Spectrom. , 2007, 21(22): 3635-3645.
[14] THINGHOLM T E, JENSEN O N, ROBINSON P J, LARSEN M R. Mol. Cell. Proteomics, 2008, 7(4): 661-671.
[15] QUAN Q, FENG J, LUI L T, SHI T, CHU I K. J. Chromatogr. A, 2017, 1498: 196-206.
[16] HUANG H, LI L, WU C, SCHIBLI D, COLWILL K, MA S, LI C, ROY P, HO K, SONGYANG Z, PAWSON T, GAO Y, LI SS. Mol. Cell. Proteomics, 2008, 7(4): 768-784.
[17] BIAN Y, LI L, DONG M, LIU X, KANEKO T, CHENG K, LIU H, VOSS C, CAO X, WANG Y, LITCHFIELD D, YE M, LIS S C, ZOU H. Nat. Chem. Biol. , 2016, 12(11): 959-966.
[18] MAKWANA M V, MUIMO R, JACKSON R F. Lab. Invest. , 2018, 98(3): 291-303.
[19] FUHRMANN J, SUBRAMANIAN V, THOMPSON P R. Angew. Chem. Int. Ed. , 2015, 54(49): 14715-14718.
[20] POTEL C M, LIN M H, HECK A J R, LEMEER S. Nat. Methods, 2018, 15(3): 187-190.
[21] HU Y, WENG Y, JIANG B, LI X, ZHANG X, ZHAO B, WU Q, LIANG Z, ZHANG L, ZHANG Y. Sci. China: Chem. ,2019, 62(6): 708-712.
[22] HU Y, JIANG B, WENG Y, SUI Z, ZHAO B, CHEN Y, LIU L, WU Q, LIANG Z, ZHANG L, ZHANG Y. Nat. Commun. ,2020, 11(1): 6226.
[23] SHI Wen-Hao, TONG Meng-Sha, LI Kai, WANG Yu-Shen, DING Chen. Prog. Biochem. Biophys. , 2018, 45(12): 1250-1258.
石文昊, 童夢莎, 李愷, 王鈺珅, 丁琛. 生物化學與生物物理進展, 2018, 45(12): 1250-1258.
[24] WELLS J M, MCLUCKEY S A. Methods Enzymol. , 2005, 402: 148-185.
[25] PAULO J A, SCHWEPPE D K. Proteomics, 2021, 21(9): e2000140.
[26] ZHANG Y, FICARRO S B, LI S, MARTO J A. J. Am. Soc. Mass Spectrom. , 2009, 20(8): 1425-1434.
[27] CUI L, REID G E. Proteomics, 2013, 13(6): 964-973.
[28] ZUBAREV R A, KELLEHER N L, MCLAFFERTY F W. J. Am. Chem. Soc. , 1998, 120, 3265-3266.
[29] KRUGER N A, ZUBAREV R A, CARPENTER B K, KELLEHER N L, HORN D M, MCLAFFERTY F W. Int. J. Mass Spectrom. , 1999, 182-183: 1-5.
[30] SYKA J E P, COON J J, SCHROEDER M J, SHABANOWITZ J, HUNT D J. Proc. Natl. Acad. Sci. U. S. A. , 2004, 101:9528-9533.
[31] POTEL C M, LEMEER S, HECK A J R. Anal. Chem. , 2019, 91(1): 126-141.
[32] FRESE C K, ALTELAAR A M, TOORN H V D, NOLTING D, GRIEP-RAMING J, HECK A J, MOHAMMED S. Anal.Chem. , 2012, 84(22): 9668-9673.
[33] FRESE C K, ZHOU H, TAUS T, ALTELAAR A M, MECHTLER K, HECK A J, MOHAMMED S. J. Proteome Res. , 2013,12(3): 1520-1525.
[34] PENKERT M, HAUSER A, HARMEL R, FIEDLER D, HACKENBERGER C P R, KRAUSE E. J. Am. Soc. Mass Spectrom. , 2019, 30(9): 1578-1585.
[35] BRODBELT J S. Chem. Soc. Rev. , 2014, 43(8): 2757-2783.
[36] FORT K L, DYACHENKO A, POTEL C M, CORRADINI E, MARINO F, BARENDREGT A, MAKAROV A A,SCHELTEMA R A, HECK A J R. Anal. Chem. , 2016, 88(4): 2303-2310.
[37] ROBINSON M R, TALIAFERRO J M, DALBY K N, BRODBELT J S. J. Proteome Res. , 2016, 15(8): 2739-2748.
[38] SUI Shao-Hui, WANG Jing-Lan, CAI Yun, QIAN Xiao-Hong. Prog. Biochem. Biophys. , 2007, 34(3): 240-245.
隋少卉, 王京蘭, 蔡耘, 錢小紅. 生物化學與生物物理進展, 2007, 34(3): 240-245.
[39] MEGGER D A, BRACHT T, MEYER H E, SITEK B. Biochim. Biophys. Acta, 2013, 1834(8): 1581-1590.
[40] ANKNEY J A, MUNEER A, CHEN X. Annu. Rev. Anal. Chem. , 2018, 11(1): 49-77.
[41] LIU J, LI J, SUN Z, DUAN Y, WANG F, WEI G, YANG J H. J. Transl. Med. , 2021, 19(1): 339.
[42] DE OLIVEIRA T M, DE LACERDA J T J G, LEITE G G F, DIAS M, MENDES M A, KASSAB P, E SILVA C G S E,JULIANO M A, FORONES N M. Clin. Biochem. , 2020, 79: 61-69.
[43] MANN M. Nat. Rev. Mol. Cell Biol. 2006, 7(12): 952-958.
[44] HOEDT E, ZHANG G, NEUBERT T A. Adv. Exp. Med. Biol. , 2019, 1140: 531-539.
[45] CHEN X, WEI S, JI Y, GUO X, YANG F. Proteomics, 2015, 15(18): 3175-3192.
[46] SSTEPATH M, ZULCH B, MAGHNOUJ A, SCHORK K, TUREWICZ M, EISENACHER M, HAHN S, SITEK B, BRACHT T. J. Proteome Res. , 2020, 19(2): 926-937.
[47] ERBER L N, LUO A, GONG Y, BEESON M, TU M, TRAN P, CHEN Y. Nutrients, 2021, 13(1): 179.
[48] HAMMAREN H M, GEISSEN E M, POTEL C M, BECK M, SAVITSKI M M. Nat. Commun. , 2022, 13(1): 7431.
[49] LLUCENA A C R, AMORIM J C, DE PAULA LIMA C V, BATISTA M, KRIEGER M A, DE GODOY L M F, MARCHINI F K. Cell Stress Chaperones, 2019, 24(5): 927-936.
[50] BELLER N C, HUMMON A B. Mol. Omics, 2022, 18(7): 579-590.
[51] WU C, BA Q, LU D, LI W, SALOVSKA B, HOU P, MUELLER T, ROSENBERGER G, GAO E, DI Y, ZHOU H,FORNASIERO E F, LIU Y. Dev. Cell, 2021, 56(1): 111-124.e6.
[52] CHEN Y Q, SHEN H, YANG R J, WAN J B. Anal. Chim. Acta, 2021, 1179: 338839.
[53] BOERSEMA P J, AYE T T, VAN VEEN T A B, HECK A J R, MOHAMMED S. Proteomics, 2008, 8(22): 4624-4632.
[54] HSU J L, HUANG S Y, CHOW N H, CHEN S H. Anal. Chem. , 2003, 75(24): 6843-6852.
[55] LIU Z, WANG Y, YAO Y, FANG Z, MIAO Q R, YE M. J. Proteomics, 2019, 208: 103501.
[56] MERTINS P, UDESHI N D, CLAUSER K R, MANI D R, PATEL J, ONG S E, JAFFE J D, CARR S A. Mol. Cell.Proteomics, 2012, 11(6): M111. 014423.
[57] MCALISTER G C, HUTTLIN E L, HAAS W, TING L, JEDRYCHOWSKI M P, ROGERS J C, KUHN K, PIKE I, GROTHE R A, BLETHROW J D, GYGI S P. Anal. Chem. , 2012, 84(17): 7469-7478.
[58] TEDFORD N C, WHITE F M, RADDING J A. Briefings Funct. Genomics Proteomics, 2008, 7(5): 383-394.
[59] XIAO W, YANG Z, YAN X, FENG L, LONG L, TU T, DENG N, CHEN W, XIAO B, LONG H, ZENG Y. Front. Neurol. ,2020, 11: 626013.
[60] XU R, CHEN Y, WANG Z, ZHANG C, DONG X, YAN Y, WANG Y, ZENG Y, CHEN P. Toxins, 2021, 13(8): 554.
[61] NOTARAS M, LODHI A, BARRIO-ALONSO E, FOORD C, RODRICK T, JONES D, FANG H, GREENING D, COLAK D.Mol. Psychiatry, 2021, 26(12): 7760-7783.
[62] FRIEDRICH C, SCHALLENBERG S, KIRCHNER M, ZIEHM M, NIQUET S, HAJI M, BEIER C, NEUDECKER J, KLAUSCHEN F, MERTINS P. Nat. Commun. , 2021, 12(1): 3576.
[63] ZOU M, WANG J, GAO J, HAN H, FANG Y. Oncol. Lett. , 2018, 15(3): 2889-2898.
[64] OGATA K, TSAI C F, ISHIHAMA Y. J. Proteome Res. , 2021, 20(8): 4193-4202.
[65] NEEDHAM E J, HINGST J R, PARKER B L, MORRISON K R, YANG G, ONSLEV J, KRISTENSEN J M, H?JLUND K,LING N X Y, OAKHILL J S, RICHTER E A, KIENS B, PETERSEN J, PEHM?LLER C, JAMES D E, WOJTASZEWSKI JF P, HUMPHREY S J. Nat. Biotechnol. , 2022, 40(4): 576-584.
[66] BUDNIK B, LEVY E, HARMANGE G, SLAVOV N. Genome Biol. , 2018, 19(1): 161.
[67] TSAI C F, OGATA K, SUGIYAMA N, ISHIHAMA Y. Cell Rep. Methods, 2022, 2(1): 100138.
[68] CHUA X Y, SALOMON A. J. Proteome Res. , 2021, 20(6): 3330-3344.
[69] STOPFER L E, CONAGE-POUGH J E, WHITE F M. Mol. Cell. Proteomics, 2021, 20: 100104.
[70] YI L, TSAI C F, DIRICE E, SWENSEN A C, CHEN J, SHI T, GRITSENKO M A, CHU R K, PIEHOWSKI P D, SMITH R D, RODLAND K D, ATKINSON M A, MATHEWS C E, KULKARNI R N, LIU T, QIAN W J. Anal. Chem. , 2019, 91(9):5794-5801.
[71] KWON Y, LEE S, PARK N, JU S, SHIN S, YOO S, LEE H, LEE C. Anal. Chem. , 2022, 94(10): 4192-4200.
[72] CHUA X Y, MENSAH T, ABALLO T, MACKINTOSH S G, EDMONDSON R D, SALOMON A R. Mol. Cell. Proteomics,2020, 19(4): 730-743.
[73] TSAI C F, WANG Y T, HSU C C, KITATA R B, CHU R K, VELICKOVIC M, ZHAO R, WILLIAMS S M, CHRISLER W B,JORGENSEN M L, MOORE R J, ZHU Y, RODLAND K D, SMITH R D, WASSERFALL C H, SHI T, LIU T. Commun.Biol. , 2023, 6(1): 70.
[74] TSAI C F, ZHANG P, SCHOLTEN D, MARTIN K, WANG Y T, ZHAO R, CHRISLER W B, PATEL D B, DOU M, JIA Y,REDUZZI C, LIU X, MOORE R J, BURNUM-JOHNSON K E, LIN M H, HSU C C, JACOBS J M, KAGAN J,SRIVASTAVA S, RODLAND K D, STEVEN WILEY H, QIAN W J, SMITH R D, ZHU Y, CRISTOFANILLI M, LIU T,LIU H, SHI T. Commun. Biol. , 2021, 4(1): 265.
[75] TSAI C F, ZHAO R, WILLIAMS S M, MOORE R J, SCHULTZ K, CHRISLER W B, PASA-TOLIC L, RODLAND K D,SMITH R D, SHI T, ZHU Y, LIU T. Mol. Cell. Proteomics, 2020, 19(5): 828-838.
[76] PRUS G, HOEGL A, WEINERT B T, CHOUDHARY C. Trends Biochem. Sci. , 2019, 44(11): 943-960.
[77] GERBER S A, RUSH J, STEMMAN O, KIRSCHNER M W, GYGI S P. Proc. Natl. Acad. Sci. U. S. A. , 2003, 100(12):6940-6945.
[78] ALSUFAYAN T A, MYERS E J, QUADE B N, BRADY C T, MARSHALL A, HAQUE N, DUFFEY M E, PARKER M D.Int. J. Mol. Sci. , 2021, 22(23): 12817.
[79] FAN Y, NIRUJOGI R S, GARRIDO A, RUIZ-MARTINEZ J, BERGARECHE-YARZA A, MONDRAGON-REZOLA E,VINAGRE-ARAGON A, CROITORU I, GOROSTIDI PAGOLA A, PATERNAIN MARKINEZ L, ALCALAY R,HICKMAN R A, DURING J, GOMES S, PRATUSEVICIUTE N, PADMANABHAN S, VALLDEORIOLA F, PEREZ SISQUES L, MALAGELADA C, XIMELIS T, MOLINA PORCEL L, MARTI M J, TOLOSA E, ALESSI D R, SAMMLER E M. Acta Neuropathol. , 2021, 142(3): 475-494.
[80] COLYER J. Ann. N. Y. Acad. Sci. , 1998, 853: 79-91.
[81] STEEN H, JEBANATHIRAJAH J A, SPRINGER M, KIRSCHNER M W. Proc. Natl. Acad. Sci. U. S. A. , 2005, 102(11):3948-3953.
[82] BEKKER-JENSEN D B, BERNHARDT O M, HOGREBE A. Nat. Commun. , 2020, 11(1): 787.
[83] OLSEN J V, VERMEULEN M, SANTAMARIA A, KUMAR C, MILLER M L, JENSEN L J, GNAD F, COX J, JENSEN T S,NIGG E A, BRUNAK S, MANN M. Sci. Signal. , 2010, 3(104): ra3.
[84] HOGREBE A, VON STECHOW L, BEKKER-JENSEN D B, WEINERT B T, KELSTRUP C D, OLSEN J V. Nat. Commun. ,2018, 9(1): 1045.
[85] HEGEMAN A D, HARMS A C, SUSSMAN M R, BUNNER A E, HARPER J F. J. Am. Soc. Mass Spectrom. , 2004, 15(5):647-653.
[86] PFLIEGER D, JUNGER M A, MüLLER M, RINNER O, LEE H, GEHRIG P M, GSTAIGER M, AEBERSOLD R. Mol.Cell. Proteomics, 2008, 7(2): 326-346.
[87] WU R, HAAS W, DEPHOURE N, HUTTLIN E L, ZHAI B, SOWA M E, GYGI S P. Nat. Methods, 2011, 8(8): 677-683.
[88] CHEN S H, LIN Y C, SHIH M K, WANG L F, LIU S S, HSU J L. Molecules, 2020, 25(22): 5316.
[89] LIM M Y, O’BRIEN J, PAULO J A, GUGI S P. J. Proteome Res. , 2017, 16(11): 4217-4226.
[90] JOHNSON H, EYERS C E, EYERS P A, BEYNON R J, GASKELL S J. J. Am. Soc. Mass Spectrom. , 2009, 20(12): 2211-2220.
[91] DOMANSKI D, MURPHY L C, BORCHERS C H. Anal. Chem. , 2010, 82(13): 5610-5620.
[92] KARAYEL O, TONELLI F, VIRREIRA WINTER S, GEYER P E, FAN Y, SAMMLER E M, ALESSI D R, STEGER M,MANN M. Mol. Cell. Proteomics, 2020, 19(9): 1546-1560.
[93] TSAI C F, KU W C, CHEN Y J, ISHIHAMA Y. Methods Mol. Biol. , 2017, 1636: 313-325.
