磁性“一步法”-液相色譜-串聯(lián)質(zhì)譜法測(cè)定水產(chǎn)品中28種有機(jī)磷農(nóng)藥殘留
2024-09-01 00:00:00金慧玲劉真真范曉民葛夢(mèng)圓王嬌王新全丁立建齊沛沛
分析化學(xué) 2024年3期
關(guān)鍵詞:方法

摘要 基于磁性納米材料建立了磁性“一步法”前處理技術(shù),結(jié)合液相色譜-串聯(lián)質(zhì)譜(LC-MS/MS)方法,實(shí)現(xiàn)了水產(chǎn)品中28 種有機(jī)磷農(nóng)藥殘留的精準(zhǔn)定量分析。針對(duì)水產(chǎn)品富含蛋白質(zhì)、脂肪和脂肪酸的特點(diǎn),選擇磁性聚合物(聚二乙烯基苯-吡咯烷酮, Fe3O4-PLS)為凈化劑,通過(guò)疏水作用、氫鍵和π-π鍵等相互協(xié)同作用高效吸附基質(zhì)干擾組分,凈化樣品。優(yōu)化了萃取溶劑類(lèi)型及用量、鹽析與除水劑、Fe3O4-PLS 和C18 用量等實(shí)驗(yàn)條件。在最佳樣品前處理?xiàng)l件下, 28 種有機(jī)磷農(nóng)藥的方法檢出限(MLOD)為0.03~0.85 μg/kg,方法定量限(MLOQ)為0.50~2.00 μg/kg;不同添加濃度水平下目標(biāo)物的平均回收率為60.1%~119.0%,相對(duì)標(biāo)準(zhǔn)偏差(RSDs)為0.8%~19.0%,方法的靈敏度和準(zhǔn)確度高。與QuEChERS 方法對(duì)比,本方法在萃取溶劑、鹽析與除水劑和凈化劑用量以及分析效率等方面均展現(xiàn)出明顯的優(yōu)勢(shì)。采用本方法檢測(cè)不同種類(lèi)水產(chǎn)品中有機(jī)磷農(nóng)藥殘留,結(jié)果表明,本方法具有良好的實(shí)用性,可為水產(chǎn)品中有機(jī)磷農(nóng)藥殘留的快速分析提供新的方法依據(jù)。
關(guān)鍵詞 Fe3O4-PLS;磁性“一步法”;液相色譜-串聯(lián)質(zhì)譜;水產(chǎn)品;有機(jī)磷農(nóng)藥
水產(chǎn)品富含人體所需的蛋白質(zhì)、不飽和脂肪酸和維生素等,是人類(lèi)飲食的重要組成部分[1]。然而,水產(chǎn)品在養(yǎng)殖過(guò)程中易受到多種污染物的污染[2-4]。有機(jī)磷農(nóng)藥廣泛應(yīng)用于農(nóng)業(yè)生產(chǎn)中,可通過(guò)地表徑流、地下水滲透、沉降和廢水排放等途徑進(jìn)入水環(huán)境[5-8];同時(shí),有機(jī)磷農(nóng)藥也應(yīng)用于水產(chǎn)養(yǎng)殖中的清塘和病蟲(chóng)害防治[9-10],殘留在水中的有機(jī)磷農(nóng)藥可通過(guò)吸入、皮膚接觸和攝食等途徑污染水產(chǎn)品[3,11],并通過(guò)食物鏈的生物放大作用在人體內(nèi)積累,對(duì)消費(fèi)者的健康構(gòu)成潛在危害[12-14]。為保障水產(chǎn)品的質(zhì)量安全和消費(fèi)者的健康,亟需建立水產(chǎn)品中有機(jī)磷農(nóng)藥殘留的快速、精準(zhǔn)分析方法。
目前,水產(chǎn)品中農(nóng)藥殘留的分析通常采用有機(jī)溶劑萃取,萃取液經(jīng)固相萃取[15]或分散固相萃取(d-SPE)[11,16]凈化后,結(jié)合色譜和質(zhì)譜實(shí)現(xiàn)準(zhǔn)確定量分析。其中, d-SPE 方法多以N-丙基乙二胺(Primarysecondary amine, PSA)、十八烷基硅烷(Octadecylsilane, C18)和石墨化炭黑(Graphitized carbon black,GCB)等商品化吸附劑,以及m-ZrO2@Fe3O4、Fe3O4-OPA 和Fe3O4-PLS 等磁性材料[17-18]為凈化劑[19-20],通過(guò)吸附去除影響目標(biāo)物回收率和準(zhǔn)確度的基質(zhì)干擾物,實(shí)現(xiàn)樣品凈化,改善了SPE 方法容易堵塞柱、耗時(shí)等問(wèn)題,具有操作簡(jiǎn)便、快速和高效等優(yōu)勢(shì),在污染物殘留分析領(lǐng)域具有較好的應(yīng)用前景。特別是以磁功能材料為凈化劑的d-SPE 方法可避免凈化過(guò)程的離心分離,提升了樣品前處理效率。然而,其萃取過(guò)程仍需要一次高速離心,而離心機(jī)的體積大,處理通量低,限制了批量樣品的分析速度。Qi 等[21]通過(guò)平衡萃取和凈化過(guò)程中樣品量的差異,將萃取和凈化過(guò)程合并為一步,并基于磁功能材料快速分離的特性,構(gòu)建了磁性“一步法”前處理技術(shù),無(wú)需離心分離,單個(gè)樣品前處理時(shí)間可縮短至5 min以內(nèi),實(shí)現(xiàn)了果蔬中多農(nóng)藥殘留的高效分析。然而,磁性“一步法”在水產(chǎn)品中有機(jī)磷農(nóng)藥殘留分析方面的研究鮮見(jiàn)報(bào)道。針對(duì)水產(chǎn)品基質(zhì)復(fù)雜,富含蛋白質(zhì)、脂肪和脂肪酸等特點(diǎn),篩選對(duì)水產(chǎn)品基質(zhì)組分具有理想吸附性能的磁功能材料對(duì)保障目標(biāo)物的精準(zhǔn)定量分析至關(guān)重要。基于親脂性二乙烯苯和親水性N-乙烯基吡咯烷酮單體制備的磁性聚合物Fe3O4-PLS[17]可通過(guò)疏水作用、氫鍵和π-π鍵等作用力協(xié)同,實(shí)現(xiàn)水產(chǎn)品中脂肪、蛋白質(zhì)和脂肪酸等基質(zhì)干擾物的吸附去除,從而達(dá)到基質(zhì)凈化和準(zhǔn)確分析目標(biāo)物的目的[22-24]。
本研究以Fe3O4-PLS 為凈化劑,系統(tǒng)考察了萃取溶劑類(lèi)型和用量、鹽析與除水劑、Fe3O4-PLS 和C18用量等關(guān)鍵因素對(duì)目標(biāo)農(nóng)藥回收率的影響,得到了水產(chǎn)品中有機(jī)磷農(nóng)藥殘留分析的最佳樣品前處理?xiàng)l件,并結(jié)合LC-MS/MS 技術(shù)構(gòu)建了水產(chǎn)品中28 種有機(jī)磷農(nóng)藥殘留的精準(zhǔn)定量分析方法。采用本方法對(duì)浙江省內(nèi)不同種類(lèi)水產(chǎn)品(魚(yú)、蝦和蟹類(lèi)等)中有機(jī)磷農(nóng)藥殘留進(jìn)行監(jiān)測(cè)分析,進(jìn)一步驗(yàn)證了本方法在實(shí)際樣品分析中的適用性。
1 實(shí)驗(yàn)部分
1.1 儀器與試劑
島津8050 三重四極桿質(zhì)譜儀和Nexera X2 超高效液相色譜儀(日本Shimadzu 公司);ALC-210.2 電子天平(感量0.01 g,瑞士Mettler Toleco 公司);BS224S 電子天平(德國(guó)IKA 公司);Vortex2 渦旋儀(德國(guó)IKA 公司);Milli-Q Integral 純水儀(美國(guó)Millipore 公司)。
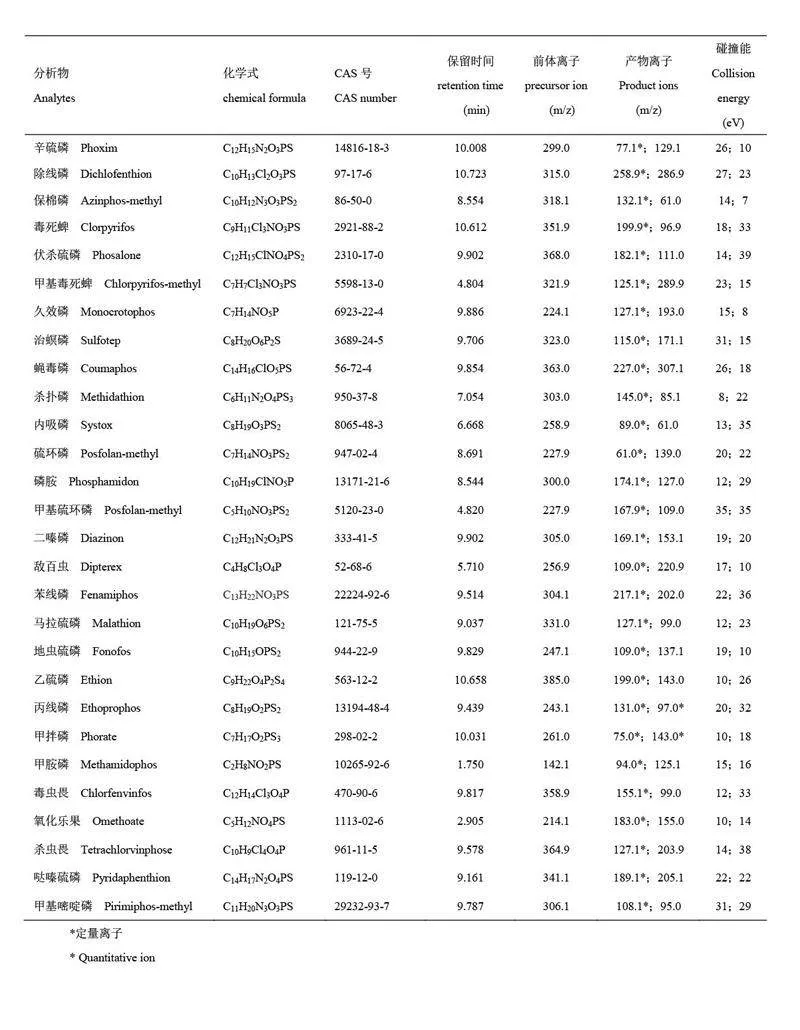
28 種有機(jī)磷農(nóng)藥標(biāo)準(zhǔn)品,純度均大于98%,購(gòu)于上海農(nóng)藥研究所和農(nóng)業(yè)部環(huán)境保護(hù)科研監(jiān)測(cè)所,詳見(jiàn)電子版文后支持信息表S1。甲醇和乙腈(色譜純,美國(guó)Merck 公司);甲酸(色譜純,美國(guó)ACS 恩科化學(xué)公司);乙酸(分析純,上海凌峰化學(xué)試劑有限公司);C18(50 μm)、PSA(40~60 μm)、NaCl 和無(wú)水MgSO4(分析純,天津博納艾杰爾科技有限公司);檸檬酸鈉和檸檬酸二鈉鹽倍半水合物(分析純,北京百靈威科技有限公司);Fe3O4-PLS(粒徑760 nm)為實(shí)驗(yàn)室自制[23]。實(shí)驗(yàn)用水為Milli-Q Integral 純水儀制備的超純水(18.2 MΩ·cm)。
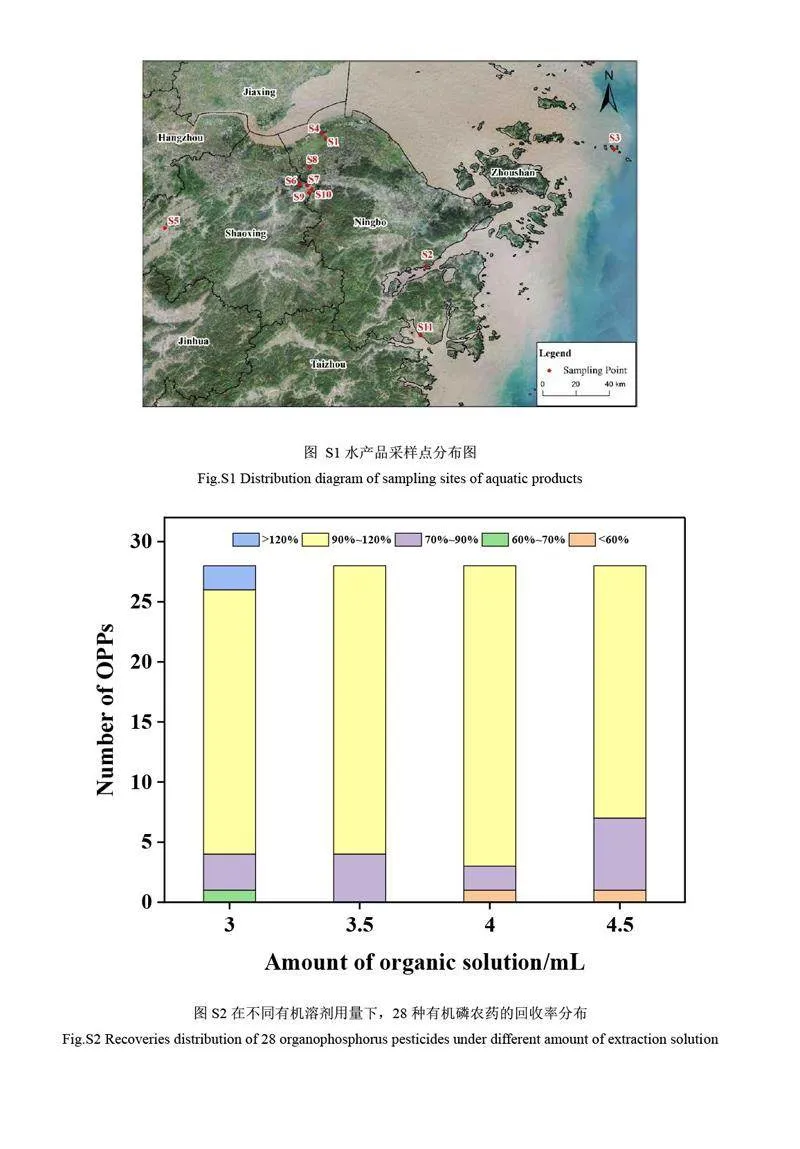
水產(chǎn)品樣品哈氏仿對(duì)蝦、梭子蟹、青蟹、南美白對(duì)蝦、美國(guó)紅魚(yú)、大黃魚(yú)、鯽魚(yú)、鱸魚(yú)、鯉魚(yú)、鯛魚(yú)和鳊魚(yú)等均購(gòu)自浙江省杭州市超市。實(shí)際樣品監(jiān)測(cè)中的18 批次水產(chǎn)品樣品分別采集于浙江省內(nèi)11 個(gè)養(yǎng)殖場(chǎng)(詳情見(jiàn)電子版文后支持信息圖S1),采樣點(diǎn)和水產(chǎn)品詳細(xì)信息見(jiàn)電子版文后支持信息表S1。水產(chǎn)品樣品經(jīng)解剖、清洗、去除其頭和尾等不可食用部位后,將肌肉組織切塊,用攪拌器攪拌均勻,于–18 ℃保存?zhèn)溆谩?/p>
1.2 實(shí)驗(yàn)方法
1.2.1 樣品前處理方法
磁性“一步法”:稱(chēng)取2.00 g(±0.02 g)上一節(jié)中制得的水產(chǎn)品樣品于50 mL 離心管中,準(zhǔn)確加入1.5 mL 水和3.5 mL 2%乙酸乙腈,渦旋1 min;加入2 g NaCl-MgSO4(1∶4, m/m)、40 mg Fe3O4-PLS 和10 mg C18,渦旋1 min, 然后將其置于長(zhǎng)方體磁鐵側(cè)面, 30 s 即可完全分離;取1 mL 上清液,經(jīng)0.22 μm濾膜過(guò)濾,進(jìn)行LC-MS/MS 分析。
QuEChERS 方法參考文獻(xiàn)[25]。準(zhǔn)確稱(chēng)取10.0 g 蝦于50 mL 離心管中,加入10 mL 乙腈,渦旋1 min,隨后加入4 g MgSO4、1 g NaCl、1 g 檸檬酸鈉和0.5 g 檸檬酸二鈉鹽倍半水合物,渦旋1 min, 4000 r/min離心5 min。準(zhǔn)確移取7 mL 上清液轉(zhuǎn)移到含有400 mg PSA、400 mg C18 和1200 mg 無(wú)水MgSO4 的15 mL離心管中,渦旋1 min, 4000 r/min 離心5 min, 上清液經(jīng)0. 22 μm 濾膜過(guò)濾,進(jìn)行LC-MS/MS 分析。
1.2.2 儀器分析條件
采用Nexera X2 超高效液相色譜儀和島津8050 三重四極桿質(zhì)譜儀進(jìn)行定量分析。采用Luna OmegaC18 色譜柱(100 mm×2.1 mm, 1.6 μm)進(jìn)行分離,柱溫箱溫度為35 ℃;流動(dòng)相A 為0.1%(V/V)甲酸溶液,流動(dòng)相B 為甲醇;梯度洗脫(0~2 min, 20% B;2~12 min, 20%~90% B;12~14 min, 90% B;14~14.2 min, 90%~20% B;14.2~20 min, 20% B);總運(yùn)行時(shí)間為20 min;流動(dòng)相流速為0.3 mL/min;進(jìn)樣體積為1 μL。
串聯(lián)質(zhì)譜采用電噴霧離子源(ESI),所有農(nóng)藥均采用正離子模式檢測(cè)。正離子模式下ESI 電壓為4000 V,毛細(xì)管溫度為300 ℃,加熱塊溫度為400 ℃, 脫溶劑管(DL)溫度為250 ℃;干燥氣(氮?dú)猓⒓訜釟猓諝猓┖挽F化氣(氮?dú)猓┑牧魉俜謩e設(shè)置為10、10 和3 L/min;碰撞氣為氬氣。串聯(lián)質(zhì)譜采用多反應(yīng)監(jiān)測(cè)(Multiple reaction monitoring, MRM)模式,每對(duì)離子的駐留時(shí)間為2 ms。每個(gè)化合物的母離子-子離子對(duì)及相應(yīng)的碰撞能量等質(zhì)譜參數(shù)詳見(jiàn)電子版文后支持信息表S2。
1.2.3 方法驗(yàn)證
配制濃度為0.5、1、2、5、10、25、50、100 和250 μg/L 的基質(zhì)標(biāo)準(zhǔn)溶液和溶劑標(biāo)準(zhǔn)溶液。通過(guò)繪制分析物的峰面積與濃度的關(guān)系擬合溶劑標(biāo)準(zhǔn)曲線和基質(zhì)匹配標(biāo)準(zhǔn)曲線,并以基質(zhì)匹配標(biāo)準(zhǔn)曲線線性方程斜率和溶劑標(biāo)準(zhǔn)曲線線性方程斜率的比值評(píng)價(jià)基質(zhì)效應(yīng)。方法檢出限(MLODs)根據(jù)3 倍信噪比和稀釋因子計(jì)算得到,方法定量限(MLOQs)根據(jù)SANTE/11312/2021 定義為滿足目標(biāo)物回收率和精密度要求的最低加標(biāo)水平。經(jīng)前處理后的樣品采用LC-MS/MS 分析。
2 結(jié)果與討論
2.1 樣品前處理方法的建立
對(duì)萃取溶劑類(lèi)型、鹽析與除水劑、Fe3O4-PLS 和C18 用量等關(guān)鍵因素進(jìn)行了優(yōu)化。選擇哈氏仿對(duì)蝦為代表基質(zhì),預(yù)先制備混配農(nóng)藥的基質(zhì)樣品,使每種有機(jī)磷農(nóng)藥的添加濃度為100 μg/kg, 經(jīng)前處理后,進(jìn)行LC-MS/MS 分析。以28 種農(nóng)藥的回收率分布情況評(píng)價(jià)總體回收率,以確定最佳的樣品前處理?xiàng)l件。
2.1.1 萃取溶劑類(lèi)型及用量的優(yōu)化
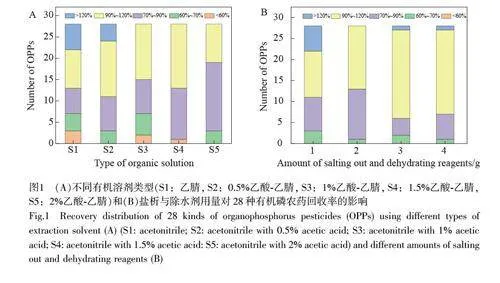
乙腈是水產(chǎn)品污染物殘留分析中兼顧目標(biāo)物萃取效率和蛋白沉淀效率的最相容溶劑[26-27],通過(guò)向乙腈中加入乙酸等溶劑可進(jìn)一步提高目標(biāo)分析物的萃取效率[28-29]。考察了不同類(lèi)型溶劑(乙腈、0.5%乙酸-乙腈、1%乙酸-乙腈、1.5%乙酸-乙腈和2%乙酸-乙腈)對(duì)28 種有機(jī)磷農(nóng)藥萃取效率的影響。結(jié)果表明(圖1A),當(dāng)萃取溶劑為純乙腈時(shí), 28 種有機(jī)磷農(nóng)藥的回收率為35.4%~189.0%,僅53.6%的有機(jī)磷農(nóng)藥回收率在70%~120%范圍內(nèi)。隨著乙酸濃度增加,有機(jī)磷農(nóng)藥回收率分布在70%~120%內(nèi)的數(shù)量逐漸增加。當(dāng)乙酸濃度增至2%時(shí), 28 種有機(jī)磷農(nóng)藥的回收率為65.2%~101.0%,其中, 89.3%的目標(biāo)物回收率分布在70%~120%范圍內(nèi)。因此,選擇2%乙酸-乙腈用于水產(chǎn)品中28 種有機(jī)磷農(nóng)藥殘留的萃取。
萃取溶劑用量也會(huì)影響有機(jī)磷農(nóng)藥回收率。探究了不同用量的2%乙酸-乙腈對(duì)28 種有機(jī)磷農(nóng)藥回收率的影響(見(jiàn)電子版文后支持信息圖S2)。當(dāng)2%乙酸-乙腈用量為3 mL 時(shí),目標(biāo)農(nóng)藥的回收率為65.8%~107.0%,其中89.3%的目標(biāo)農(nóng)藥回收率在70%~120%范圍內(nèi)。當(dāng)2%乙酸-乙腈用量為3.5 mL 時(shí),28 種有機(jī)磷農(nóng)藥的回收率為71.6%~112.0%。然而,隨著乙腈用量的繼續(xù)增加,甲胺磷和氧化樂(lè)果等農(nóng)藥回收率呈下降趨勢(shì),當(dāng)2%乙酸-乙腈用量為4.0 和4.5 mL 時(shí),目標(biāo)農(nóng)藥回收率分別為53.2%~118.0%和51.7%~117.0%。綜上,選擇2%乙酸乙腈的用量為3.5 mL。
2.1.2 鹽析和除水劑用量的優(yōu)化
NaCl 和無(wú)水MgSO4(1∶4, m/m)是常用的鹽析與除水劑組合,可促進(jìn)目標(biāo)物在有機(jī)相中的分布,提高目標(biāo)物的回收率[30-31]。考察了不同用量(1、2、3 和4 g)的NaCl 和無(wú)水MgSO4(1∶4, m/m)對(duì)28 種有機(jī)磷農(nóng)藥回收率的影響,目標(biāo)農(nóng)藥的回收率見(jiàn)圖1B。當(dāng)鹽析與除水劑用量為1 g 時(shí), 28 種農(nóng)藥的回收率為65.8%~169.0%,其中,伏殺硫磷的回收率高達(dá)169%;當(dāng)鹽析與除水劑用量增至2 g 時(shí), 28 種農(nóng)藥的回收率為67.1%~118.0%,符合分析要求;當(dāng)鹽析與除水劑用量為3.0 和4.0 g 時(shí),目標(biāo)農(nóng)藥回收率為63.0%~133.0%和64.3%~126.0%,伏殺硫磷和甲胺磷等農(nóng)藥回收率偏高(gt;120%)。因此,本研究選擇鹽析與除水劑用量為2.0 g。
2.1.3 Fe3O4-PLS用量的優(yōu)化
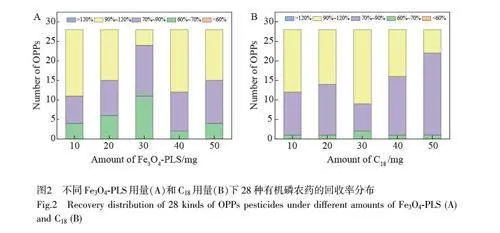
考察了不同用量(10、20、30、40 和50 mg)的凈化材料Fe3O4-PLS 對(duì)28 種有機(jī)磷農(nóng)藥回收率的影響,目標(biāo)農(nóng)藥的回收率分布見(jiàn)圖2A。不同F(xiàn)e3O4-PLS 用量下,目標(biāo)農(nóng)藥的回收率均在60.7%~104.0%內(nèi),符合分析要求。其中,當(dāng)Fe3O4-PLS 用量為40 mg 時(shí),回收率處于70%~120%的目標(biāo)農(nóng)藥數(shù)量最多,占比高達(dá)92.8%。當(dāng)Fe3O4-PLS 用量增至50 mg,目標(biāo)農(nóng)藥回收率為61.8%~98.2%,其中85.7%的目標(biāo)農(nóng)藥回收率在70%~120%內(nèi),目標(biāo)物的回收率未得到進(jìn)一步改善。本實(shí)驗(yàn)選擇Fe3O4-PLS 的用量為40 mg。
2.1.4 C18用量的優(yōu)化
C18 常用于脂肪、脂肪酸和甾醇等非極性物質(zhì)的吸附去除[32]。考察了Fe3O4-PLS(40 mg)和不同用量(10、20、30、40 和50 mg)C18 組合凈化對(duì)有機(jī)磷農(nóng)藥回收率的影響。如圖2B 所示,在不同的C18 用量下,目標(biāo)農(nóng)藥的回收率影響相當(dāng),符合殘留分析要求。考慮到節(jié)約成本,選擇C18 用量為10 mg。
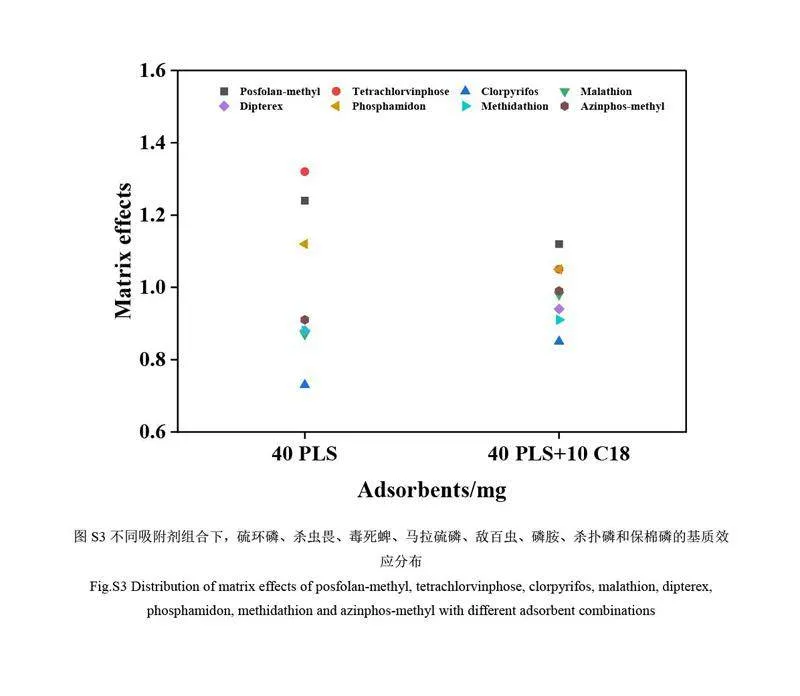
與單獨(dú)使用Fe3O4-PLS 凈化相比,萃取液經(jīng)40 mg Fe3O4-PLS 和10 mg C18 共同凈化可實(shí)現(xiàn)基質(zhì)干擾物的高效吸附去除,凈化效果較佳,尤其是硫環(huán)磷、殺蟲(chóng)畏、毒死蜱、馬拉硫磷、敵百蟲(chóng)、磷胺、殺撲磷和保棉磷等目標(biāo)物的基質(zhì)效應(yīng)明顯減弱(電子版文后支持信息圖S3)。
2.2 方法驗(yàn)證
2.2.1 線性范圍、方法檢出限及方法定量限
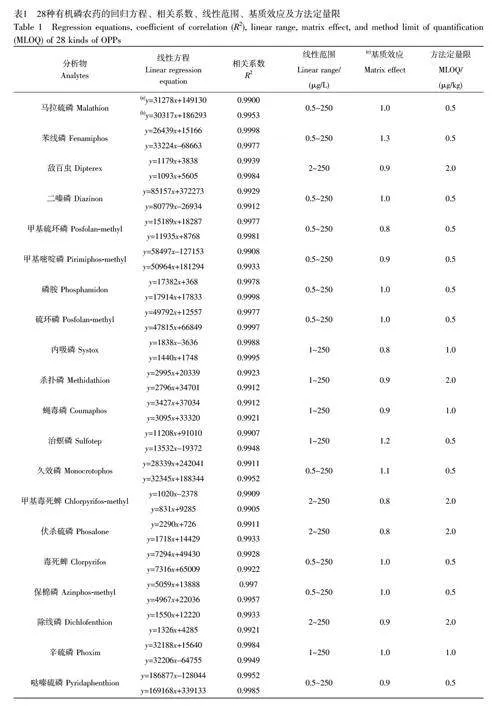
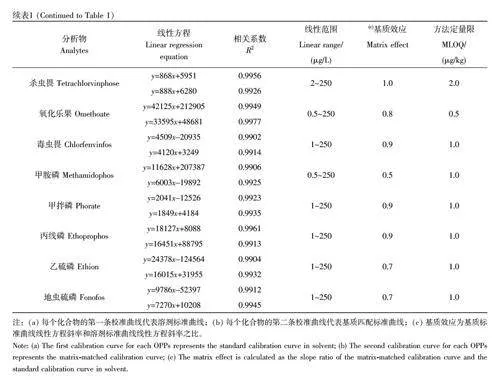
配制濃度為0.5、1、2、5、10、25、50、100 和250 μg/L 的基質(zhì)標(biāo)準(zhǔn)溶液和溶劑標(biāo)準(zhǔn)溶液。其中,溶劑標(biāo)準(zhǔn)溶液采用甲醇溶液配制而成,基質(zhì)標(biāo)準(zhǔn)溶液采用空白基質(zhì)的提取溶液配制。溶液經(jīng)LC-MS/MS分析后,通過(guò)分析物的峰面積(y)與濃度(x,μg/L)的關(guān)系擬合溶劑標(biāo)準(zhǔn)曲線和基質(zhì)匹配標(biāo)準(zhǔn)曲線方程,線性方程、基質(zhì)效應(yīng)和MLOD 等參數(shù)見(jiàn)表1。二嗪磷、馬拉硫磷和苯線磷等13 種目標(biāo)農(nóng)藥的線性范圍為0.5~250 μg/L;內(nèi)吸磷、甲胺磷和地蟲(chóng)硫磷等9 種目標(biāo)農(nóng)藥在1~250 μg/L 范圍內(nèi)線性關(guān)系良好;敵百蟲(chóng)、殺撲磷和殺蟲(chóng)畏等6 種目標(biāo)農(nóng)藥在2~250 μg/L 范圍內(nèi)具有良好的線性關(guān)系。28 種農(nóng)藥的MLODs為0.03~0.85 μg/kg, MLOQs 為0.5~2.0 μg/kg。
2.2.2 基質(zhì)效應(yīng)
基質(zhì)效應(yīng)通過(guò)基質(zhì)標(biāo)準(zhǔn)曲線線性方程斜率和溶劑標(biāo)準(zhǔn)曲線線性方程斜率的比值進(jìn)行評(píng)價(jià)[33],其中,當(dāng)斜率比為1 時(shí),表示無(wú)基質(zhì)效應(yīng);當(dāng)斜率比小于1 時(shí),呈基質(zhì)抑制效應(yīng);當(dāng)斜率比大于1 時(shí),呈基質(zhì)增強(qiáng)效應(yīng)。同時(shí),根據(jù)基質(zhì)效應(yīng)的差異程度,進(jìn)一步對(duì)基質(zhì)/溶劑標(biāo)準(zhǔn)溶液線性的斜率比值進(jìn)行細(xì)化。當(dāng)斜率比在0.8~1.2 之間時(shí),基質(zhì)效應(yīng)抑制或增強(qiáng)效果較弱;斜率比在0.5~0.8 或1.2~1.5 范圍內(nèi),表現(xiàn)為中等基質(zhì)效應(yīng);當(dāng)斜率比小于0.5 或大于1.5 時(shí),表示基質(zhì)效應(yīng)增強(qiáng)或抑制效果較為顯著[34]。由表1 可見(jiàn),28 種目標(biāo)農(nóng)藥的斜率比為0.51~1.26;其中,僅有苯線磷、甲胺磷、乙硫磷和地蟲(chóng)硫磷4 種農(nóng)藥表現(xiàn)出明顯的基質(zhì)效應(yīng),表明基質(zhì)效應(yīng)對(duì)目標(biāo)物的定量分析影響相對(duì)較小。為了實(shí)現(xiàn)所有分析目標(biāo)物的準(zhǔn)確定量分析,仍采用基質(zhì)匹配標(biāo)準(zhǔn)溶液曲線進(jìn)行濃度校正。
2.2.3 回收率和精密度
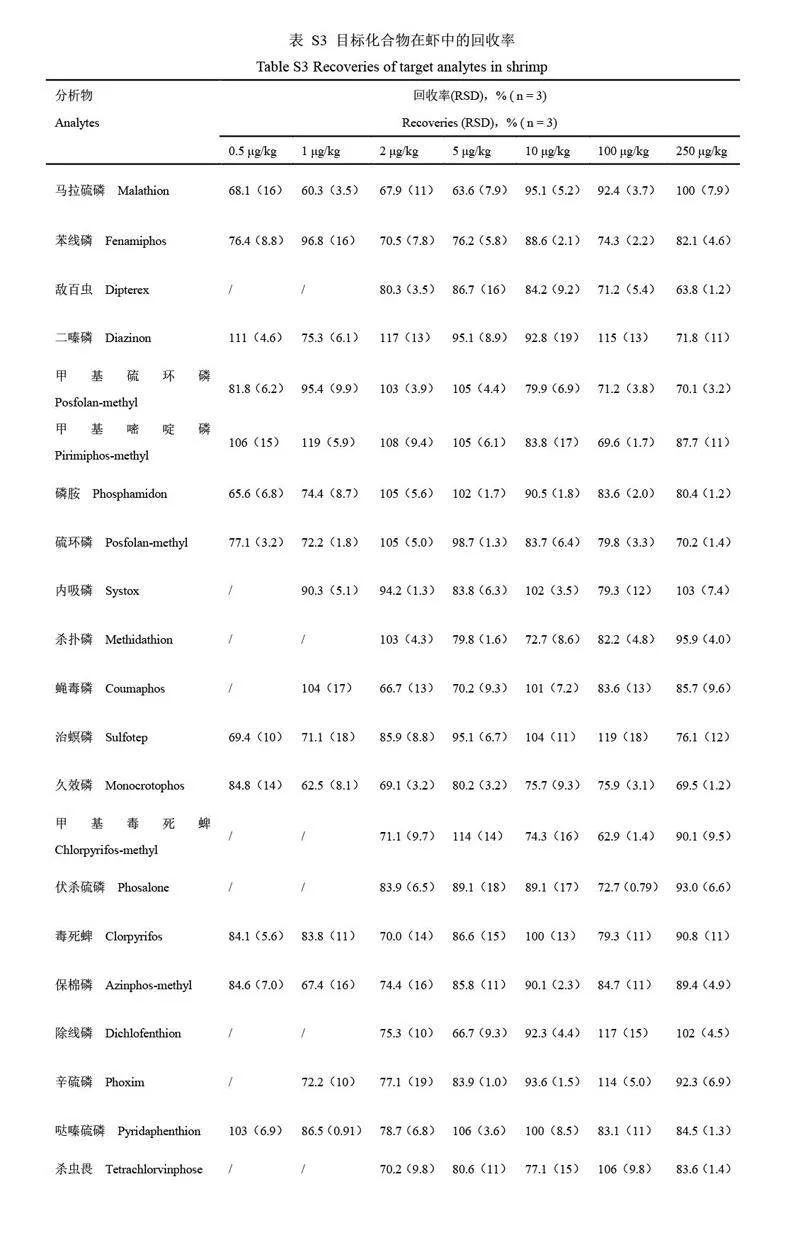
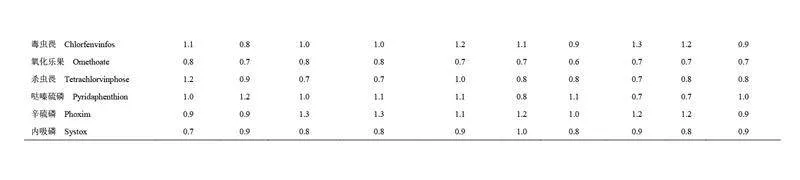
選取空白蝦樣品基質(zhì)進(jìn)行添加回收實(shí)驗(yàn),目標(biāo)物添加濃度水平分別為0.5、1、2、5、10、100 和250 μg/kg(n=3),按照1.2.1 節(jié)的方法進(jìn)行檢測(cè),目標(biāo)農(nóng)藥的回收率見(jiàn)電子版文后支持信息表S3。馬拉硫磷、苯線磷和甲基硫環(huán)磷等13 種目標(biāo)農(nóng)藥在0.5~250 μg/kg 添加水平下的回收率為60.3%~119.0%,RSDs 為0.9%~19.0%。受儀器靈敏度限制,內(nèi)吸磷、蠅毒磷和地蟲(chóng)硫磷等9 種目標(biāo)農(nóng)藥在添加濃度為0.5 μg/kg 時(shí)未檢出,當(dāng)目標(biāo)物添加濃度為1~250 μg/kg 時(shí),該9 種目標(biāo)農(nóng)藥的回收率為60.1%~118.0%,RSDs 為0.8%~19.0%;敵百蟲(chóng)、殺撲磷和伏殺硫磷等6 種目標(biāo)農(nóng)藥在添加濃度為0.5 和1.0 μg/kg 時(shí)未檢出,當(dāng)添加濃度2~250 μg/kg 時(shí),回收率為62.9%~117.0%, RSDs 為1.2%~18.0%。綜上,在0.5~250 μg/kg 的添加水平下, 28 種目標(biāo)農(nóng)藥回收率為60.1%~119.0%, RSDs 為0.8%~19.0%,方法的準(zhǔn)確度和精密度符合殘留分析要求。
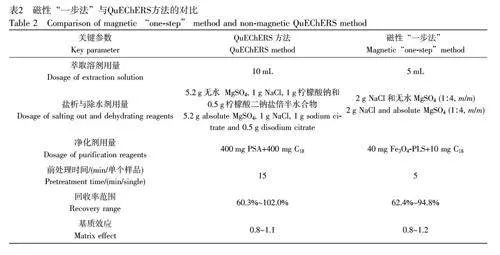
2.2.4 本方法與QuEChERS方法對(duì)比
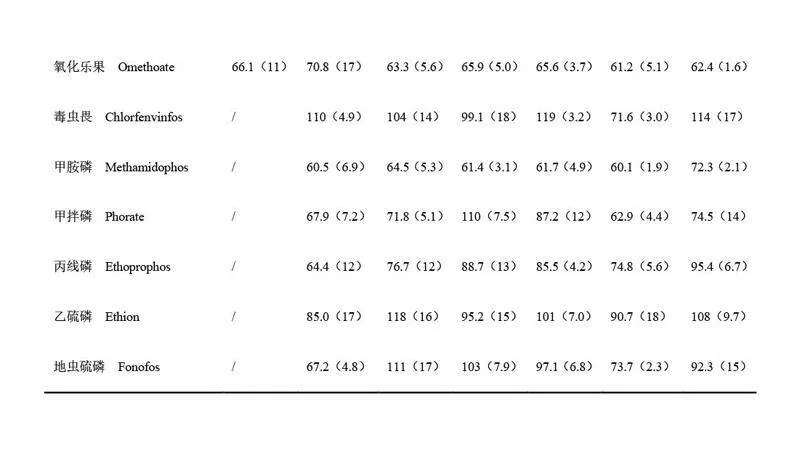
將磁性“一步法”與QuEChERS 方法在萃取溶劑、鹽析與除水劑和凈化劑等用量、樣品前處理時(shí)間以及目標(biāo)物回收率等方面進(jìn)行對(duì)比(表2)。選取空白蝦樣品基質(zhì)進(jìn)行添加回收實(shí)驗(yàn),添加濃度水平為100 μg/kg(n=3)。QuEChERS 方法和本方法的目標(biāo)物回收率分別為60.3%~102.0%和62.4%~94.8%,均滿足農(nóng)藥殘留分析要求。為實(shí)現(xiàn)目標(biāo)物的理想萃取效率, QuEChERS 方法中單個(gè)樣品萃取溶劑的消耗量為10 mL, 本方法中單個(gè)樣品的萃取溶劑用量為5 mL, 僅為QuEChERS 方法的1/2。在鹽析與除水劑用量方面, QuEChERS 方法需消耗7.7 g 鹽析與除水劑,本方法的鹽析與除水劑用量?jī)H為2 g。在樣品凈化過(guò)程中, QuEChERS 方法中凈化劑的用量為400 mg PSA 和400 mg C18,本方法僅需使用40 mg Fe3O4-PLS和10 mg C18 即可實(shí)現(xiàn)樣品的理想凈化。更重要的是, QuEChERS 方法的單個(gè)樣品前處理時(shí)間約為15 min, 而本方法在5 min 之內(nèi)即可完成樣品前處理,大大縮短了樣品前處理時(shí)間,顯著提高了樣品前處理效率。本方法在萃取溶劑、鹽析與除水劑和凈化劑用量以及樣品前處理時(shí)間等方面均展現(xiàn)出明顯優(yōu)勢(shì)。
2.2.5 不同水產(chǎn)品中的驗(yàn)證研究
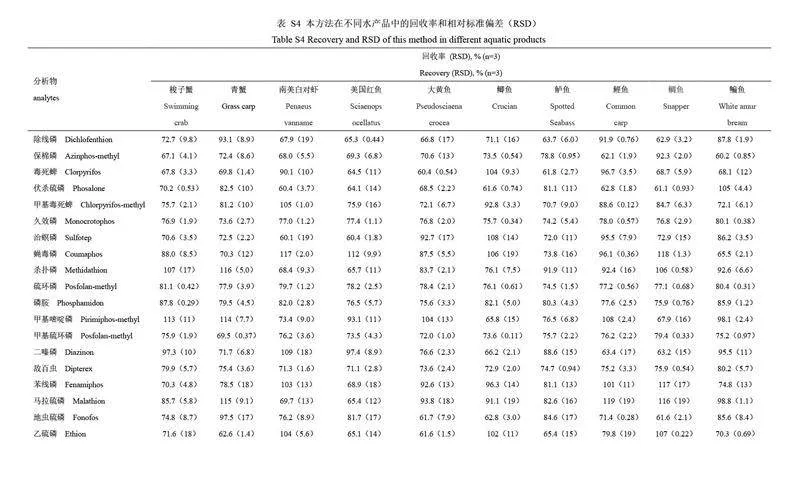
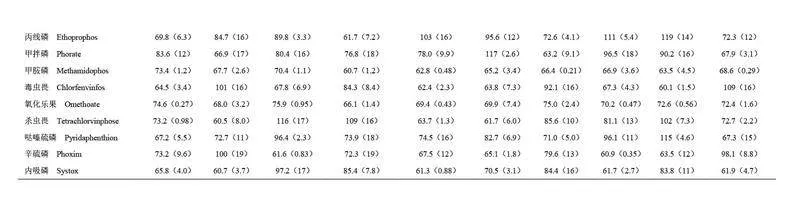
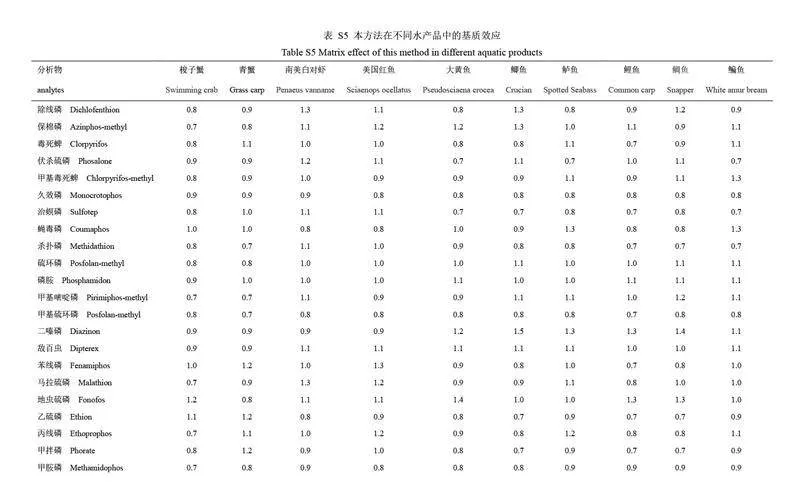
為評(píng)價(jià)本方法對(duì)不同水產(chǎn)品中有機(jī)磷農(nóng)藥殘留分析的適用性,選取10 種不同種類(lèi)的水產(chǎn)品空白樣品(梭子蟹、青蟹、南美白對(duì)蝦、美國(guó)紅魚(yú)、大黃魚(yú)、鯽魚(yú)、鱸魚(yú)、鯉魚(yú)、鯛魚(yú)和鳊魚(yú))進(jìn)行回收率驗(yàn)證實(shí)驗(yàn)。目標(biāo)農(nóng)藥的添加濃度為100 μg/kg(n=3),不同水產(chǎn)品中目標(biāo)農(nóng)藥的回收率結(jié)果和基質(zhì)效應(yīng)見(jiàn)電子版文后支持信息表S4 和S5。結(jié)果表明,雖然目標(biāo)農(nóng)藥的回收率因水產(chǎn)品種類(lèi)和目標(biāo)農(nóng)藥化學(xué)性質(zhì)的不同而存在差異,但所有水產(chǎn)品中的目標(biāo)農(nóng)藥回收率均在60%~120%范圍內(nèi), RSD 均小于20%,符合農(nóng)藥殘留分析要求。同時(shí), 28 種有機(jī)磷農(nóng)藥在不同種類(lèi)的水產(chǎn)品中的基質(zhì)效應(yīng)范圍為0.6~1.4,其中79.3%的有機(jī)磷農(nóng)藥未表現(xiàn)出明顯的基質(zhì)效應(yīng)。因此,本方法在不同種類(lèi)水產(chǎn)品有機(jī)磷農(nóng)藥殘留分析方面具有良好的適用性。
2.2.6 實(shí)際樣品分析
采用本方法對(duì)浙江省內(nèi)養(yǎng)殖場(chǎng)中18 種水產(chǎn)品中的有機(jī)磷農(nóng)藥殘留進(jìn)行分析,目標(biāo)水產(chǎn)品包括白條魚(yú)(2)、鯉魚(yú)(3)、草魚(yú)(2)、鯰魚(yú)(1)、黑魚(yú)(1)、鯽魚(yú)(2)、七星鱸(1)、大嘴黑鱸(1)、美國(guó)紅魚(yú)(1)、大黃魚(yú)(3)和青蟹(1),具體采樣信息見(jiàn)電子版文后支持信息圖S1 和表S2。結(jié)果表明,僅在鯰魚(yú)中檢出毒死蜱,檢出濃度為0.54 μg/kg, 其余27 種有機(jī)磷農(nóng)藥在所有水產(chǎn)品中均未檢出。目前,國(guó)內(nèi)外缺乏水產(chǎn)品中目標(biāo)農(nóng)藥的最大殘留限量標(biāo)準(zhǔn),然而,根據(jù)我國(guó)國(guó)家標(biāo)準(zhǔn)(GB 27632021)和歐盟文件(396/2005)中制定的農(nóng)產(chǎn)品中有機(jī)磷農(nóng)藥殘留的最大殘留限量(0.01~10 mg/kg),水產(chǎn)品中檢測(cè)到的毒死蜱濃度較低,處在可安全食用范圍內(nèi)。
3 結(jié)論
本研究基于磁性Fe3O4-PLS 為凈化劑,開(kāi)發(fā)了磁性“一步法”前處理技術(shù),結(jié)合LC-MS/MS,建立了水產(chǎn)品中28 種有機(jī)磷農(nóng)藥殘留的精準(zhǔn)定量分析方法。考察了萃取溶劑類(lèi)型、鹽析與除水劑、Fe3O4-PLS 和C18 用量等對(duì)目標(biāo)農(nóng)藥回收率的影響,獲得最佳前處理?xiàng)l件。28 種有機(jī)磷農(nóng)藥的MLODs 為0.03~0.85 μg/kg, MLOQs 為0.50~2.00 μg/kg。不同添加濃度水平下,目標(biāo)物的回收率在60.1%~119.0%范圍內(nèi), RSDs 為0.8%~19.0%,符合殘留分析要求。與QuEChERS 方法相比,磁性“一步法”在萃取溶劑、鹽析與除水劑、凈化劑用量等方面展現(xiàn)出明顯的優(yōu)勢(shì),尤其在提升樣品前處理效率方面,可將單個(gè)樣品前處理時(shí)間縮短至5 min 內(nèi)。進(jìn)行了磁性“一步法”在不同種類(lèi)水產(chǎn)品中有機(jī)磷農(nóng)藥殘留分析的驗(yàn)證研究,結(jié)果表明,本方法在不同種類(lèi)水產(chǎn)品有機(jī)磷農(nóng)藥殘留分析中具有較好的適用性,可為水產(chǎn)品中有機(jī)磷農(nóng)藥殘留監(jiān)管、隱患排查、毒理效應(yīng)及風(fēng)險(xiǎn)評(píng)估等研究提供可靠的技術(shù)支撐。
References
[1] MELENCHON F, LARRAN A M, SANZ M A, RICO D, FABRIKOV D, BARROSO F G, GALAFAT A, ALARCON F J,MORALES A E, HIDALGO M C, LOUREN?O H M, PESSOA M F, TOMAS-ALMENAR C. Fishes, 2023, 8(6): 286.
[2] JIMOH J O, RAHMAH S, MAZELAN S, JALILAH M, OLASUNKANMI J B, LIM L S, GHAFFAR M A, CHANG Y M,BHUBALAN K, LIEW H J. Environ. Pollut. , 2023, 317: 120769.
[3] HU S L, CHEN X C, XUE Y X, ZHI L Y, YANG Y H, ZHU Y G, XUE X M. Exposure Health, 2023, 16(1): 57-70.
[4] GU L, HU B, FU Y, ZHOU W, LI X, HUANG K, ZHANG Q, FU J, ZHANG H, ZHANG A, FU J, JIANG G. Water Res. ,2023, 240: 120083.
[5] KAUSHAL J, KHATRI M, ARYA S K. Ecotoxicol. Environ. Saf. , 2021, 207: 111483.
[6] QUAGLIA G, JORIS I, BROEKX S, DESMET N, KOOPMANS K, VANDAELE K, SEUNTJENS P. J. Environ. Manage. ,2019, 246: 583-593.
[7] UDDIN M H, SHAHJAHAN M, AMIN A R, HAQUE M M, ISLAM M A, AZIM M E. Aquacult. Rep. , 2016, 3: 88-92.
[8] WANG S, YANG G, TANG Y, WANG Y, SHEN X, SI W, YU H, ZHAI W, FODJO E K, KONG C. Foods, 2023, 12(6):1131.
[9] SHI Q, YANG H, CHEN Y, ZHENG N, LI X, WANG X, DING W, ZHANG B. Int. J. Mol. Sci. , 2023, 24(13): 11099.
[10] ZHANG L, LIU Y, CHEN H, CAI W. Comp. Biochem. Physiol. , Part C: Toxicol. Pharmacol. , 2022, 257: 109333.
[11] LI J, ZHAO L, LETCHER R J, ZHANG Y, JIAN K, ZHANG J, SU G. Environ. Int. , 2019, 127: 35-51.
[12] LAO Y M, QU C L, ZHANG B, JIN H. Food Chem. , 2023, 402: 134500.
[13] AJIBOYE T O, OLADOYE P O, OLANREWAJU C A, AKINSOLA G O. Environ. Nanotechnol. , Monit. Manage. , 2022,17: 100655.
[14] YU X, XU F, ZHANG R, LIU H, SUN A, ZHANG L, ZHANG Z, SHI X. Sci. Total Environ. , 2023, 883: 163633.
[15] WANG S, HUANG X, WANG M, TIAN L, LI X, KONG C, HAN F, LOU X, YE H, SHI Y. Anal. Lett. , 2022, 55(5): 784-795.
[16] TONG Xue-Zhi, CHEN Dong-Yang, FENG Jia-Li, FAN Xiang, ZHANG Hao, YANG Sheng-Yuan. Chin. J. Chromatogr,2023, 41(6): 490-496.
童學(xué)智, 陳東洋, 馮家力, 范翔, 張昊, 楊勝園. 色譜, 2023, 41(6): 490-496.
[17] LIU Z, ZHAO H, WANG J, WANG Z, DI S, XU H, WANG Q, WANG X, WANG X, QI P. Ecotoxicol. Environ. Saf. , 2022,241: 113830.
[18] PENG X T, JIANG L, GONG Y, HU X Z, PENG L J, FENG Y Q. Talanta, 2015, 132: 118-125.
[19] ZHANG L, MA J, LIU P, QI A, JIN H, JIA R, ZHENG Z, YAN C, CAI M. Front. Mar. Sci. , 2023, 10: 1167712.
[20] KIM K, CHOI Y, MOK S, MOON H B, JEON J. Food Chem. , 2023, 399: 133958.
[21] QI P, WANG J, LIU Z, WANG Z, XU H, DI S, ZHAO H, WANG X. J. Chromatogr. A, 2021, 1659: 462589.
[22] HE Z, WANG P, LIU D, ZHOU Z. Talanta, 2014, 127: 1-8.
[23] LIU Z, QI P, WANG J, WANG Z, DI S, XU H, ZHAO H, WANG Q, WANG X, WANG X. Sci. Total Environ. , 2020, 708:135221.
[24] YAO Z, JIAO W, SHAO F, SONG H, ZHANG H, ZHOU Q, LI A. Chem. Eng. J. , 2019, 360: 511-518.
[25] BARBIERI M V, POSTIGO C, GUILLEM-ARGILES N, MONLLOR-ALCARAZ L S, SIMIONATO J I, STELLA E,BARCELO D, ALDA M L D. Sci. Total Environ. , 2019, 653: 958-967.
[26] SHIN D, KIM J, KANG H S. Food Control, 2021, 120: 107552.
[27] VARENINA I, BILANDZIC N, LUBURIC D B, KOLANOVIC B S, VARGA I. Food Control, 2022, 133: 108576.
[28] LI S, MENG Z, LIU Y, LIU D, XU Z. J. Food Compos. Anal. , 2022, 113: 104712.
[29] PERESTRELO R, SILVA P, PORTO-FIGUEIRA P, PEREIRA J A M, SILVA C, MEDINA S, CAMARA J S. Anal. Chim.Acta, 2019, 1070: 1-28.
[30] ZHANG C, DENG Y, ZHENG J, ZHANG Y, YANG L, LIAO C, SU L, ZHOU Y, GONG D, CHEN L, LUO A. TrAC, Trends Anal. Chem. , 2019, 118: 517-537.
[31] KIM L, LEE D, CHO H K, CHOI S D. Trends Environ. Anal. Chem. , 2019, 22: e00063.
[32] LONG Y, HUANG Y, ZHU M, MA Y, GAN B, WANG Y X, YU Q, XIE J, CHEN Y. Food Chem. , 2023, 409: 135265.
[33] PRODHAN M, AFROZE M, BEGUM A, AHMED M, SARKER D. Int. J. Environ. Anal. Chem. , 2023, 103(6): 1292-1303.
[34] WANG S, QI P, DI S, WANG J, WU S, WANG X, WANG Z, WANG Q, WANG X, ZHAO C, LI Q. Anal. Chim. Acta, 2019,1074: 108-116.
猜你喜歡
中老年保健(2021年9期)2021-08-24 03:52:04
河北畫(huà)報(bào)(2021年2期)2021-05-25 02:07:46
中學(xué)生數(shù)理化(高中版.高考理化)(2020年2期)2020-04-21 05:33:04
兒童繪本(2020年5期)2020-04-07 17:46:30
兒童故事畫(huà)報(bào)(2019年5期)2019-05-26 14:26:14
Coco薇(2016年2期)2016-03-22 02:42:52
山東青年(2016年1期)2016-02-28 14:25:23
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長(zhǎng)指南(2015年7期)2015-08-11 15:03:12
小雪花·成長(zhǎng)指南(2015年4期)2015-05-19 14:47:56
