多色免疫組織化學技術在腫瘤標志物分析中的研究進展
2024-09-01 00:00:00楊馬駿楊超杰賀月趙燦冀海偉王琦秦玉嶺吳麗
分析化學 2024年3期

摘要 多色免疫組織化學(Multiplex immunohistochemistry, mIHC)技術是一種新型多靶點病理組織染色、成像及分析技術。該技術通過在單張組織切片上檢測多種生物標志物的表達水平及空間分布,實現對細胞的表型、組成、形態及細胞間相互作用機理的全面解析。近年來, mIHC 技術被成功應用于腫瘤免疫研究領域,為腫瘤的診斷和治療開辟了新的途徑。本文從近年開發的mIHC 檢測技術出發,針對不同類型mIHC技術的檢測原理及其特點進行了詳細闡述,重點討論了新型mIHC 檢測技術在腫瘤標志物及腫瘤微環境檢測等領域中的應用,并對mIHC 在腫瘤免疫診斷和治療領域的應用前景和發展趨勢進行了展望。
關鍵詞 多色免疫組織化學;熒光多色標記;腫瘤診斷;循環熒光成像;評述
2018 年諾貝爾生理學和醫學獎分別授予了美國得州大學免疫學家詹姆斯·艾利森(James P Allison)教授和日本京都大學本庶佑(Tasuku Honjo)教授,以表彰他們發現“負性免疫調節”治療癌癥的療法。這徹底顛覆了以往人類對抗腫瘤的策略,標志著腫瘤免疫治療進入了一個新的歷史階段。目前上市的腫瘤免疫治療藥物雖然效果很好,但是存在價格高、整體應答率較低以及部分患者的不良反應較大等缺點。以免疫檢查點PD-1/PD-L1 單抗為例,其在腫瘤治療中的臨床整體應答率僅有20%~30%,無法實現有針對性的靶向用藥,造成醫療資源浪費,限制了其在腫瘤治療中的應用[1-3]。腫瘤免疫微環境(Tumorimmune microenvironment, TIME)生物標志物與免疫治療反應息息相關。因此,治療腫瘤的關鍵是制定針對不同病人的個體化腫瘤精準免疫治療方案。然而。由于TIME 呈現明顯腫瘤異質性,而且面臨多種細胞類型并存、各種細胞因子相互作用以及免疫抑制的復雜狀況,依賴于對單一標志物的檢測難以全面、深入和精準地描繪TIME 的全貌[4-9]。為了實現腫瘤精準免疫治療,亟需開發高效且精準的免疫組織化學(免疫組化)技術用于臨床免疫診斷中腫瘤標志物及TIME 的精準檢測[10-12]。
目前,采用多色免疫組織化學(Multiplex immunohistochemistry, mIHC)技術對免疫組織化學標志物及腫瘤微環境檢測的總結性文章已有報導,如Tan 等[13]介紹了幾種已經臨床應用的mIHC 技術,詳細比較了mIHC 聯合熒光免疫檢測法(Immunofluorescence, IF)、芯片細胞術、基于DNA 條形碼的mIHC/IF、多重IHC/IF 分析方案以及三維(3D)成像等mIHC 技術在免疫組織化學(Immunohistochemistry, IHC)標志物檢測中的差異。但是,該文并沒有對一些新開發的檢測技術進行介紹。Wilson 等[14]對通過mIHC 研究TIME 時的數據預處理、質量控制和分析方法等方面進行了綜述,側重于分析過程和數據處理的介紹。Hoyt 等[15]介紹了多重免疫熒光技術(Multiplex immunofluorescence, mIF)的原理、技術方法以及檢測數據的優化分析過程,并探討了其臨床應用潛力。迄今為止,針對最新開發的mIHC 及其創新點、局限性和應用潛力等方面的綜述性文章還未見報道。本文詳細闡述了近十年發展起來的mIHC 技術的檢測原理、創新點以及不足之處,重點討論了這些檢測技術在免疫組織化學腫瘤標志物檢測等領域中的應用,并展望了新型mIHC 技術在腫瘤免疫診斷中的發展前景和趨勢。
1 IHC技術
傳統的IHC 技術是應用免疫學抗原-抗體反應,對組織或細胞內抗原或抗體等物質進行定性分析和定位的技術,是描述腫瘤微環境空間組學信息的常見手段,在臨床病理診斷中應用廣泛。目前,發展較成熟的IHC 技術是使用酶或熒光標記的抗體對福爾馬林固定且石蠟包埋的樣本(Formalin-fixed andparrffin-embedded, FFPE)進行染色。該方法能標記1~3 種抗體,但不能完整地反映IHC 腫瘤標志物的種類及進行TIME 的檢測,導致患者錯失可能的精準化免疫治療機會。雖然該方法可采用多種抗體標記及連續切片技術提高對多種組分和多個方位的檢測能力,但連續切片并不能在完全相同的細胞層面進行解析,增加了漏檢機率,還會丟失空間位置信息,不利于待檢蛋白質的精準定位,費時費力且需消耗大量的樣本。
2 mIHC技術
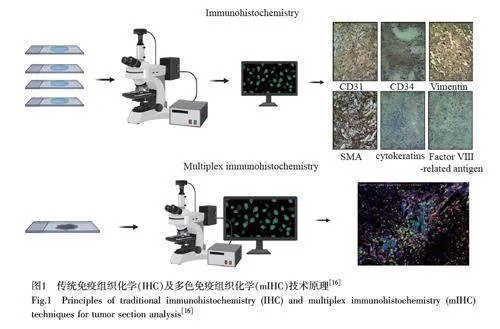
近年來,隨著IHC 及檢測儀器的發展,研究者開發了mIHC 技術。mIHC 是利用免疫學和細胞化學的原理,在同一張切片上同時或先后采用不同顏色的熒光素或酶促產物原位顯示組織或細胞內不同大分子抗原物質的一項多標記染色技術(圖1)[16]。目前,該技術可在一張組織切片上同時完成8~20 種甚至更多種類IHC 標志物的檢測,大大提高了抗原分型和篩選的效率。mIHC 可直接評估腫瘤和TIME 中多種細胞的表型及空間關系,精確反映TIME 的免疫狀態,不僅能節省檢測時間、提高對稀有組織標本的利用率,更能幫助研究人員全面、深入和精準地了解腫瘤局部組織和微環境信息[17-18]。根據mIHC 的檢測結果可以準確了解TIME 中腫瘤抗原表達和生長特征、浸潤性免疫細胞的數量、表型和功能狀態以及細胞因子種類和表達量,并依此制定針對特定腫瘤的免疫治療方案[19-20]。目前,多表位配體法[21]、連續免疫過氧化物酶標記和清除法[22]、循環免疫熒光法[23]、多重免疫熒光法[24]、酪胺信號放大法[25]、交換反應信號放大法[26]、數字空間分析法[27]和質譜流式細胞成像[28]等新技術被應用到mIHC 研究中,大大推動了mIHC 技術的發展,使得mIHC 成為腫瘤精準免疫治療、單細胞測序及TIME 研究的重要手段,具有廣闊的臨床應用前景[19-20]。
2.1 多表位配體技術
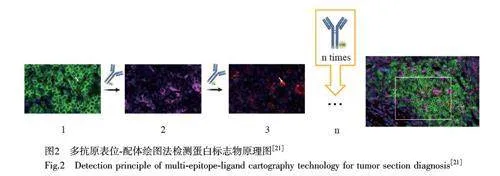
常用的商業化染料(如eFluor 和AmCyan 等)的熒光發射譜較寬,染料之間有光譜重疊,導致可同時識別的熒光探針數量有限。為提高分析通量,研究人員發展了多種基于不同熒光信號移除技術的多色循環免疫熒光成像方法。多表位配體法(Multiepitope ligand cartography, MELC)是一種基于高通量熒光顯微鏡實現對亞細胞水平蛋白標志物識別和量化的方法。MELC 首先采用熒光標記抗體選擇性結合組織樣品中的蛋白標志物,通過熒光成像實現對蛋白標志物空間位置的選擇性標記,得到熒光圖像后,通過光漂白去除熒光標記物,再應用另一組標記抗體對相應分子定位成像[16]。重復多次該步驟可得到多種蛋白標志物分子定位圖(圖2)[21]。Eckhardte 等[29]運用該技術標記單一組織切片中的主要目標細胞,檢測淋巴或骨髓中各種細胞活性及分化狀態。該方法有望用于研究轉基因小鼠自身免疫性腦脊髓炎病變或評估人類多發性硬化癥治療潛在靶點的藥物反應性。但是,該方法用于檢測鑒別不同細胞類型時,存在難以保持抗體染色質量的一致性和檢測有效性的問題。
為了進一步拓展MELC 在蛋白標志物檢測中的應用范圍, Holzarth 等[30]使用MELC 闡明和量化基質細胞間的異質性,實現了對基質細胞和造血細胞的多重空間分析。該研究成功使用多達100 個熒光標記物對同一組織切片中的蛋白標志物進行染色,并進一步通過圖像處理和圖像精確疊加分析了蛋白標志物的表達。此外, MELC 還被用于繪制固定細胞或組織中的多種蛋白質圖譜[21,31]。然而,逐個區域光漂白掃描操作效率較低、耗時長,還存在殘余背景信號干擾等問題。此外,易被光漂白的熒光分子并不適用于熒光成像研究,而光學穩定性強的熒光標簽又難被光漂白,該矛盾制約了MELC 技術的廣泛使用。
2.2 連續免疫過氧化物酶標記和清除技術
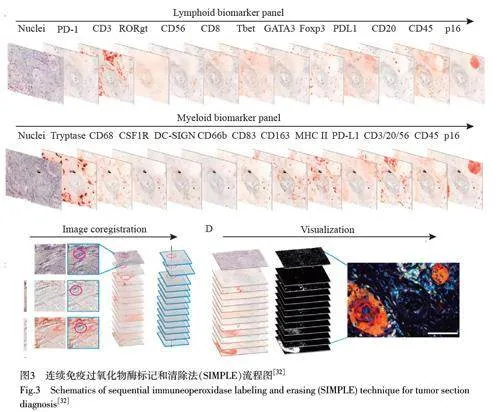
弗吉尼亞大學生物醫學工程系的Glass等[22]開發了一種連續免疫過氧化物酶標記和清除法(Sequentialimmuneoperoxidase labeling and erasing, SIMPLE)。采用該技術可同時檢測多種抗原,利用醇溶性過氧化物酶底物3-氨基-9-乙基咔唑(9-Ethylcarbazol-3-amine, AEC)標記抗原分子, AEC 與抗原分子結合并在有機溶劑中產生紅色沉淀;清除沉淀,抗體在酸化的高錳酸鹽中洗脫,再進行下一輪染色,達到多重標記的目的(圖3)[32]。在小鼠或人的FFPE 組織切片中進行多抗原共定位時,使用AEC 組織染色成像后,在乙醇中去除AEC 沉淀,再在酸化的KMnO4 溶液(0.15 mol/L KMnO4, 0.01 mol/L H2SO4)中洗脫抗體,對組織進行下一輪染色,在1 個組織切片中可同時顯示至少5 種蛋白標記物。該研究采用包含38 例頭頸部鱗狀細胞癌的組織芯片揭示了淋巴、髓系細胞密度和人類乳頭狀瘤病毒載量與患者預后情況密切相關。SIMPLE 法的優勢是在組織樣本數量有限的條件下能重復多次對相同細胞或組織中的多種抗原進行分析,不足之處在于使用酸化的KMnO4 洗脫液除去結合的抗體時可能破壞組織完整性,導致局部組織退化、成像質量降低,并且只局限于5 個熒光循環通道,檢測時間較長[22,32]。
2.3 循環免疫熒光技術
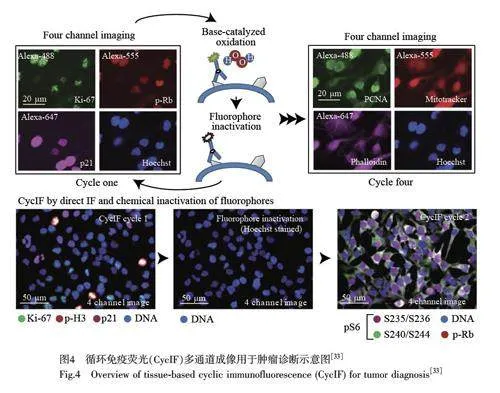
循環免疫熒光技術(Cyclic immunofluorescence, CycIF)是一種使用傳統熒光顯微鏡進行多重免疫熒光成像的方法,將常見熒光標記物與抗體結合,通過連續的4~6 通道組織成像獲得相應蛋白標志物的成像圖。該方法使用H2O2 氧化熒光標記物來淬滅熒光團,實現對抗體的標記-淬滅-再標記過程,可構建多達30 個通道的熒光成像圖。與抗體標記剝離方法不同, CycIF 的反應條件較溫和且易于標準化。美國哈佛醫學院的Sorger 研究組[33]使用多種抗體固定和標記COLO858 黑色素瘤細胞,通過標記-淬滅-再標記的循環免疫熒光法構建了15 通道連續熒光成像,在同一組織中實現了多種蛋白標志物的檢測(圖4)。
然而,該方法存在熒光團失活、信號不足、染色不均勻以及細胞易丟失等問題。在CycIF 的基礎上, Sorger 研究組[34]進一步發展了組織循環免疫熒光法(Tissue-based cyclic immunofluorescence, t-CycIF),對固定在載玻片上的FFPE 標本進行多重免疫熒光成像。t-CycIF 可對不同正常組織和腫瘤組織中的信號轉導級聯、腫瘤抗原和免疫標記物進行定量分析。與CycIF 不同的是, t-CycIF 解決了CycIF 無法在同一切片上檢測蛋白標志物的難題,可標記來自人體的正常和腫瘤組織樣本中的60 余種蛋白質。與其它IHC 方法相比, CycIF 使用易標準化的檢測試劑和儀器,價格低廉,檢測成本低于傳統的免疫熒光法,因此更適用于高通量分析和篩選。但是, t-CycIF 存在檢測細胞易丟失、循環通道少和分析周期長等問題[23,35]。
2.4 多重免疫熒光技術
在單個組織標本中僅通過顯微鏡表征蛋白質的結構和DNA 分子的數量限制了對健康和疾病生物學基礎的理解。Gerdes 等[36]建立了多重免疫熒光法(Multiplexed immunofluorescence, MxIF),可在FFPE 中對多種分析物進行定量分析,并對單細胞或亞細胞中的蛋白標志物進行表征。該方法首先在熒光染料結合抗體標記組織前采集組織背景熒光,待染料淬滅后,使用新的抗體重新標記組織獲取新的熒光成像圖,重復多次后可以實現對細胞和亞細胞多靶點的定量分析。利用此方法對747 例結直腸癌患者的61 種蛋白標識物的原位成像及共定位進行分析,深度揭示了腫瘤的異質性。MxIF 的技術特點是開發了滅活熒光溶液,可在15 min 內有效消除菁基染料熒光。實驗結果表明,經滅活熒光溶液處理5 min 后, Cy3 和Cy5 的熒光強度分別降低了60%和80%;處理15 min 后,熒光信號強度降低到初始強度的2%以下。MxIF 的實驗結果表明,可檢測分析物的數量不止60 種,并未達到該方法的檢測上限。總體而言,該技術克服了目前熒光顯微成像技術的一些局限性,解決了標準免疫熒光方法中隨著分析物數量的增加對細胞的定量檢測、共定位等分子特征難以進行分析的難題,此外,該方法還可應用于腫瘤以外的組織或細胞成像研究[24,37-38]。但是, MxIF 采用H2O2 洗脫以清除熒光信號的方法,存在耗時長且抗原表位易損傷的風險,仍然有很大的改進空間。
2.5 酪胺信號放大技術
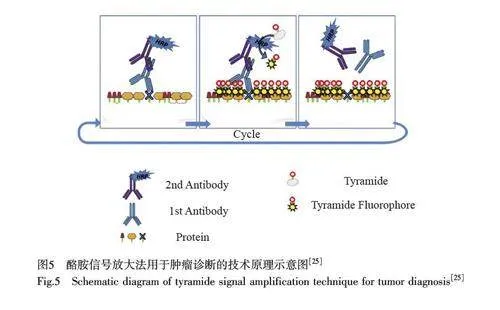
酪胺信號放大技術(Tyramide signal amplification, TSA)是一類利用辣根過氧化物酶(Horseradishperoxidase, HRP)對靶蛋白或核酸進行高密度原位標記的檢測方法。TSA 方法的原理是利用酪胺鹽在HRP 催化和H2O2 作用下形成共價鍵結合位點,產生大量的酶促產物[39-40]。該產物可與周圍的蛋白殘基(包括色氨酸、組氨酸和酪氨酸殘基)結合,從而在抗原-抗體結合部位產生大量的生物素沉積,再與隨后加入的鏈霉親和素-HRP/熒光基團結合,經多次循環放大,可以結合大量的酶分子或熒光基團[41-42],使其檢測信號得到幾何級放大(圖5)[25]。Pienta 研究組[41]利用TSA 對轉移結腸癌患者腫瘤微環境中的細胞進行鑒別,系統分析了細胞表型和免疫細胞的浸潤性,深入了解疾病成因,為臨床決策提供了參考。
TSA 中每個反應體系僅有單一抗體,因此不存在抗體交叉以及一抗、二抗種屬的匹配問題,克服了實驗設計時不同種屬抗體選擇的限制[43]。此外, TSA 將流式細胞術與IHC 獲得的信息相結合,通過在一個組織切片上進行高通量、高分辨率識別和準確的圖像分析,能夠揭示各種細胞類型之間的關系,這使得TSA 在腫瘤免疫治療領域、探索腫瘤細胞與其微環境之間的相互作用和設計組合治療方法等方面具有良好的應用潛力。同時, TSA 技術具有極強的敏感度,可檢測極微量的目的抗原,并且能極大地降低抗體的用量。但是,該方法最多只能同時分析7 種抗體,分析單個樣本需要15~20 h, 僅染色標記就需要6~8 h,并且無法避免非特異性信號的干擾[25,44]。
2.6 交換反應信號放大技術
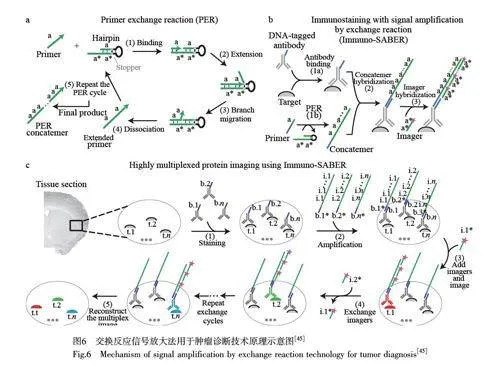
熒光原位雜交技術(Fluorescence in situ hybridization, FISH)是將熒光基團連接到探針上,利用熒光標記的特定核酸序列與細胞內相應的靶DNA 或RNA 分子雜交,通過在熒光顯微鏡下觀察熒光信號來確定與探針雜交后被染色的細胞或細胞器的形態和分布,或觀察結合了熒光探針的DNA 或RNA 分子在染色體及其它細胞器中定位的方法。在FISH 的基礎上,為了解決高效采樣和連續檢測的問題,研究者開發了一種新的方法即交換反應信號放大法(Signal amplification by exchange reaction, SABER)(圖6)[45]。
SABER 技術通過DNA 條形碼標記的抗體和引物交換反應產生的正交DNA 結合物實現高度多重信號放大,可以同時分析十幾種靶標物質。SABER 法的基本原理是DNA 的兩條核酸鏈之間可以通過堿基互補配對形成DNA 雙鏈,通過設計合適的輔助探針即可使整條鏈不斷地自組裝雜交連接形成長鏈的DNA 串聯體,此DNA 串聯體可作為信號放大的載體。與傳統的FISH 技術相比, SABER 的信號放大是獨立可編碼的,無需原位酶參與反應,通過與多個帶有熒光團的核酸結合以實現信號放大。該方法在組織檢測中具有獨立調節、高效靶向的優點。哈佛大學的Kishi 等[46]使用該方法在固定的腫瘤細胞和腫瘤組織中將RNA 和DNA 的FISH 信號放大5~450 倍,將SABER 與膨脹顯微鏡相結合,實現了快速、多通道和高分辨率組織成像,為蛋白質的擴增成像提供了一個高效的平臺。該技術是通過12~16 種靶向標記物實現檢測信號放大的引物交換反應,并且過程高度可控。然而,該方法需要大量核酸標記的抗體,操作復雜、耗時長且檢測費用較高[26]。
2.7 數字空間分析技術
數字空間分析技術(Digital spatial profiling, DSP)是一種對FFPE 樣品中蛋白質或RNA 多重空間分析的方法[27]。DSP 是將現代基因組檢測技術、光化學和微流體采樣相結合,將經典的IHC 和原位雜交技術結合發展而成的可進行基因組和蛋白質組多重解析分析的集成系統[47]。Beechem 研究組[47]使用該方法分析了淋巴組織、免疫組織、結腸和直腸腫瘤中多達44 種蛋白質和96 個基因的空間分布。DSP平臺的工作原理如圖7 所示,寡核苷酸鏈通過對紫外光敏感且可光裂解的連接基團與抗體或RNA 雜交探針偶聯。將抗體或探針與石蠟包埋的組織共同孵育,使之與蛋白質或RNA 充分結合后,使用可編程數字微鏡設備引導紫外光精確照射所檢測的區域(Region of interest, ROI),將寡核苷酸標簽與抗體解偶聯,樣本可再次使用。最后采用nCounter 儀器對寡核苷酸條形碼進行計數和數字定量,形成組織形態的數字化圖像。該研究組已對約1400 個基因和5000 個探針進行了復雜的數字信號處理和基因圖譜測試,并且檢測數量上還有提升空間。DSP 平臺和nCounter 平臺結合,可同時對40 多種蛋白質和5000 種RNA 進行分析。該方法對組織樣本的傷害極小,臨床樣本經DSP 分析后不僅可再次用于其它分析,還可長期保存,以便后續的溯源或用于后續研究分析。該方法也可基于熒光標記自動對組織中的稀有細胞類型、任意形狀或尺寸的區域進行分析,甚至可對單個細胞的蛋白質表達水平進行定量分析,為臨床患者的診斷和預后提供依據。2020 年, Farren 等[48]將nCounter 與DSP 技術聯用,以胰腺癌FFPE 樣本為研究對象,針對4 種臨床治療策略選取了24 例樣本,首先利用nCounter 表達譜技術探尋不同治療方法之間基因表達的差異,再通過生物信息學技術對基因組、蛋白質種類、分子復合物以及EnrichR 分析啟動子進行分析,并采用DSP 技術分析了胰腺癌微環境中各種免疫相關蛋白標記物的數量和空間分布在不同治療方法中的變化。DSP 技術將表達譜的多重定量信息與組織原位信息整合,可在1 張石蠟組織切片上實現多達上百種蛋白和上千種mRNA 的原位共分析,將組織病理學、腫瘤免疫與表達譜完美結合,在腫瘤微環境研究、腫瘤異質性與腫瘤免疫研究中有顯著優勢。然而, DSP 只能獲取目標區域的多重信息,缺少圖像信息,對細胞的空間分布只能提供有限的信息,并且DSP 單次分析區域較小、光照效率低以及耗時較長的缺點限制了其應用范圍。
DSP 與SABER 這兩種方法是利用核酸堿基相互組合產生的數以萬計的編碼區分不同抗體及其對應的蛋白。DSP 和SABER 可一步標記需要觀測的所有抗體,并對各個抗體逐一進行核酸解碼識別。此外,還可采用循環熒光成像的方式,通過核酸互補競爭結合或核酶降解的方式移除帶有熒光信號的編碼核酸,實現重復免疫熒光染色。這兩種核酸編碼的方法較前面幾種信號移除方法更溫和有效,但成本和消耗時間方面并不具有優勢。
2.8 質譜流式細胞成像技術
質譜流式細胞成像技術(Imaging mass cytometry, IMC)是一種已商業化的成像技術,其中代表性工作之一是Nolan 研究組[49]關于質譜細胞儀(Mass cytometry, CyTOF)的研究。CyTOF 通過重金屬同位素標記抗體或探針,并利用質譜細胞儀對標記樣品進行檢測,得到了詳細的細胞表面或內部的信號數據。該技術對單細胞表型和功能性蛋白的研究具有重要意義。CyTOF 技術繼承了傳統流式細胞技術的高速分析能力,同時還具備質譜檢測的高分辨率特性,已成為流式細胞技術領域的一個全新發展方向。與傳統的流式細胞技術相比,該方法具有上百個相對獨立的分析通道,可準確鑒別不同原子質量的金屬同位素,并具備多樣化的數據處理方式,可對樣品進行深入分析[28]。
在CyTOF 研究基礎上, Nolan 研究組[50]進一步發展了多重離子束成像技術(Multiplexed ion beamimaging, MIBI)。該技術首先使用40~100 種不同金屬標記的抗體對腫瘤組織切片染色,金屬標記物被高能量束電離成次級離子,次級離子被送入質譜成像儀進行檢測。以MIBItracker 軟件作為組織樣本成像的分析工具,實現了腫瘤切片中多種標志物的可視化共定位分析,彌補了CyTOF 技術在分析細胞表型與空間位置關系中應用的局限。其中, CyTOF 和MIBI 都已有商業化儀器,可同時分析100 種腫瘤標志物,并具有較高的靈敏度。但是,該方法需要制備用于質譜檢測的標記抗體,在檢測過程中需要額外使用流式細胞儀,并且只能進行小區域的樣本分析,因此不能很好地對整個切片做出客觀評價。此外,該方法對檢測樣本中的細胞邊界界定不清晰,成像圖中單個像素可能包含不止1 個細胞的信號。隨著免疫診斷腫瘤標志物的不斷發現,該方法的診斷時間逐漸延長,因此這類技術不適合在臨床上大規模應用。
3 總結與展望
在腫瘤免疫治療的時代,雖然傳統的IHC/IF 仍是檢測腫瘤標志物的主要手段,但受到組織樣本有限、染色過程冗長費力、數據采集及分析系統落后等因素的限制。隨著對腫瘤發病機理的認識逐漸深刻,迫切需要實施腫瘤精準醫療。mIHC 成像技術不僅可實現特定的多種蛋白標志物的檢測,得到細胞組成和空間排列的全面信息,還可反映腫瘤微環境中的免疫狀態,在臨床檢測和轉化醫學領域具有強大的應用潛力。未來,還需要對mIHC 成像技術進行更多的探索,以及嚴格的大規模驗證研究。
References
[1] STACK E C, FOUKAS P G, LEE P P. J. Immunother. Cancer, 2016, 4(1): 9.
[2] LU S, STEIN J E, RIMM D L, WANG D W, BELL J M, JOHNSON D B, SOSMAN J A, SCHALPER K A, ANDERS R A,WANG H, HOYT C, PARDOLL D M, DANILOVA L, TAUBE J M. JAMA Oncol. , 2019, 5(8): 1195-1204.
[3] YEONG J, TAN T, CHOW Z L, CHENG Q, LEE B, SEET A, LIM J X, LIM J C T, ONG C C H, THIKE A A, SARAF S,TAN B Y C, POH Y C, YEE S, LIU J, LIM E, IQBAL J, DENT R, TAN P H. J. Clin. Pathol. , 2020, 73(9): 557-562.
[4] PETRICOIN E F, BELLUCO C, ARAUJO R P, LIOTTA L A. Nat. Rev. Cancer, 2006, 6(12): 961-967.
[5] NILSSON J, SKOG J, NORDSTRAND A, BARANOV V, MINCHEVA-NILSSON L, BREAKEFIELD X O, WIDMARK A.Br. J. Cancer, 2009, 100(10): 1603-1607.
[6] JONES A, DHANAPALA L, KANKANAMAGE R N T, KUMAR C V, RUSLING J F. Anal. Chem. , 2020, 92(1): 345-362.
[7] PRENSNER J R, RUBIN M A, WEI J T, CHINNAIYAN A M. Sci. Transl. Med. , 2012, 4(127): 127rv3.
[8] CORCORAN R B, CHABNER B A. N. Engl. J. Med. , 2018, 379(18): 1754-1765.
[9] FARES J, FARES M Y, KHACHFE H H, SALHAB H A, FARES Y. Signal Transduction Targeted Ther. , 2020, 5(1): 28.
[10] LI Zhong, SUN Yan, QIAN Qi-Jun. Chin. J. Cancer Biother. , 2019, 26(1): 7-15.
李忠, 孫艷, 錢其軍. 中國腫瘤生物治療雜志, 2019, 26(1): 7-15.
[11] WU Si-Wen, LIU Bao-Rui. Chin. J. Cancer Biother. , 2021, 28(8): 769-774.
吳思雯, 劉寶瑞. 中國腫瘤生物治療雜志, 2021, 28(8): 769-774.
[12] FERLAY J, COLOMBET M, SOERJOMATARAM I, PARKIN D M, PI?EROS M, ZNAOR A, BRAY F. Int. J. Cancer,2021, 149(4): 778-789.
[13] TAN W C C, NERURKAR S N, CAI H Y, NG H H M, WU D, WEE Y T F, LIM J C T, YEONG J, LIM T K H. Cancer Commun. , 2020, 40(4): 135-153.
[14] WILSON C M, OSPINA O E, TOWNSEND M K, NGUYEN J, SEGURA C M, SCHILDKRAUT J M, TWOROGER S S,PERES L C, FRIDLEY B L. Cancers, 2021, 13(12): 3031.
[15] HOYT C C. Front. Mol. Biosci. , 2021, 8: 674747.
[16] WIDODO S S, HUTCHINSON R A, FANG Y, MANGIOLA S, NEESON P J, DARCY P K, BARROW A D, HOVENS C M,DINEVSKA M, STYLLI S S, MANTAMADIOTIS T. Cancer Immunol. Immunother. , 2021, 70(7): 1811-1820.
[17] ZHANG W, SONG Z J, ZHANG B Y, WANG J L, GUO Q, SUN Z W, TANG H. Neoplasma, 2021, 68(6): 1272-1282.
[18] TAUBE J M, ROMAN K, ENGLE E L, WANG C, BALLESTEROS-MERINO C, JENSEN S M, MCGUIRE J, JIANG M,COLTHARP C, REMENIUK B, WISTUBA I, LOCKE D, PARRA E R, FOX B A, RIMM D L, HOYT C. J. Immunother.Cancer, 2021, 9(7): e002197.
[19] CAPALBO C, SCAFETTA G, FILETTI M, MARCHETTI P, BARTOLAZZI A. Int. J. Mol. Sci. , 2019, 20(7): 1607.
[20] GOLTSEV Y, SAMUSIK N, KENNEDY-DARLING J, BHATE S, HALE M, VAZQUEZ G, BLACK S, NOLAN G P. Cell,2018, 174(4): 968-981.e15.
[21] BONNEKOH B, POMMER A J, B?CKELMANN R, PHILIPSEN L, HOFMEISTER H, GOLLNICK H. J. Dtsch. Dermatol.Ges. , 2008, 6(12): 1038-1051.
[22] GLASS G, PAPIN J A, MANDELL J W. J. Histochem. Cytochem. , 2009, 57(10): 899-905.
[23] ENG J, THIBAULT G, LUOH S W, GRAY J W, CHANG Y H, CHIN K. Methods Mol. Biol. , 2020, 2055: 521-562.
[24] FRANCISCO-CRUZ A, PARRA E R, TETZLAFF M T. Methods Mol. Biol. , 2020, 2055: 467-495.
[25] STACK E C, WANG C, ROMAN K A, HOYT C C. Methods, 2014, 70(1): 46-58.
[26] LIN K, TOMHON P, LEHMKUHL S, LAASNER R, THEIS T, BLUM V. ChemPhysChem, 2021, 22(19): 1947-1957.
[27] SMITH K D, PRINCE D K, HENRIKSEN K J, NICOSIA R F, ALPERS C E, AKILESH S. Kidney Int. , 2022, 101(5):1017-1026.
[28] CATENA R, MONTUENGA L M, BODENMILLER B. J. Pathol. , 2018, 244(4): 479-484.
[29] ECKHARDT J, OSTALECKI C, KUCZERA K, SCHULER G, POMMER A J, LECHMANN M. J. Histochem. Cytochem. ,2013, 61(2): 125-133.
[30] HOLZWARTH K, KOHLER R, PHILIPSEN L, TOKOYODA K, LADYHINA V, WAHLBY C, NIESNER R A, HAUSER A E. Cytometry, Part A, 2018, 93A(9): 876-888.
[31] PIERRE S, SCHOLICH K. Mol. Biosyst. , 2010, 6(4): 641-647.
[32] TSUJIKAWA T, KUMAR S, BORKAR R N, AZIMI V, THIBAULT G, CHANG Y H, BALTER A, KAWASHIMA R, CHOE G, SAUER D, EL RASSI E, CLAYBURGH D R, KULESZ-MARTIN M F, LUTZ E R, ZHENG L, JAFFEE E M,LEYSHOCK P, MARGOLIN A A, MORI M, GRAY J W, FLINT P W, COUSSENS L M. Cell Rep. , 2017, 19(1): 203-217.
[33] LIN J R, FALLAHI-SICHANI M, SORGER P K. Nat. Commun. , 2015, 6(1): 8390.
[34] LIN J R, IZAR B, WANG S, YAPP C, MEI S, SHAH P M, SANTAGATA S, SORGER P K. eLife, 2018, 7: e31657.
[35] LIN J R, FALLAHI-SICHANI M, CHEN J Y, SORGER P K. Curr. Protoc. Chem. Biol. , 2016, 8(4): 251-264.
[36] GERDES M J, SEVINSKY C J, SOOD A, ADAK S, BELLO M O, BORDWELL A, CAN A, CORWIN A, DINN S, FILKINS R J, HOLLMAN D, KAMATH V, KAANUMALLE S, KENNY K, LARSEN M, LAZARE M, LI Q, LOWES C, MCCULLOCH C C, MCDONOUGH E, MONTALTO M C, PANG Z, RITTSCHER J, SANTAMARIA-PANG A,SARACHAN B D, SEEL M L, SEPPO A, SHAIKH K, SUI Y, ZHANG J, GINTY F. Proc. Natl. Acad. Sci. U.S.A. , 2013,110(29): 11982-11987.
[37] NELSON D A, MANHARDT C, KAMATH V, SUI Y, SANTAMARIA-PANG A, CAN A, BELLO M, CORWIN A, DINN S R, LAZARE M, GERVAIS E M, SEQUEIRA S J, PETERS S B, GINTY F, GERDES M J, LARSEN M. Biol. Open, 2013,2(5): 439-447.
[38] ENNINGA E A L, LEONTOVICH A A, FEDYSHYN B, WAKEFIELD L, GANDHI M, MARKOVIC S N, RUANO R,KERR S E. Reprod. Sci. , 2020, 27(5): 1129-1138.
[39] AKAMA K, SHIRAI K, SUZUKI S. Anal. Chem. , 2016, 88(14): 7123-7129.
[40] ZHANG S, HU B, XIA X, XU Y, HANG B, JIANG J, HU J. Molecules, 2019, 24(23): 4364.
[41] ROY S, AXELROD H D, VALKENBURG K C, AMEND S, PIENTA K J. J. Cell. Biochem. , 2019, 120(4): 4804-4812.
[42] BERNACKI K D, FIELDS K L, ROH M H. Diagn. Cytopathol. , 2014, 42(7): 570-575.
[43] TAUBE J M, AKTURK G, ANGELO M, ENGLE E L, GNJATIC S, GREENBAUM S, GREENWALD N F, HEDVAT C V,HOLLMANN T J, JUCO J, PARRA E R, REBELATTO M C, RIMM D L, RODRIGUEZ-CANALES J, SCHALPER K A,STACK E C, FERREIRA C S, KORSKI K, LAKO A, RODIG S J, SCHENCK E, STEELE K E, SURACE M J, TETZLAFF M T, VON LOGA K, WISTUBA I I, BIFULCO C B. J. Immunother. Cancer, 2020, 8(1): e000155.
[44] BERENS M E, SOOD A, BARNHOLTZ-SLOAN J S, GRAF J F, CHO S, KIM S, KIEFER J, BYRON S A, HALPERIN R F,NASSER S, ADKINS J, CUYUGAN L, DEVINE K, OSTROM Q, COUCE M, WOLANSKY L, MCDONOUGH E,SCHYBERG S, DINN S, SLOAN A E, PRADOS M, PHILLIPS J J, NELSON S J, LIANG W S, AL-KOFAHI Y, RUSU M,ZAVODSZKY M I, GINTY F. PLoS One, 2019, 14(12): e0219724.
[45] SAKA S K, WANG Y, KISHI J Y, ZHU A, ZENG Y, XIE W, KIRLI K, YAPP C, CICCONET M, BELIVEAU B J, LAPAN S W, YIN S, LIN M, BOYDEN E S, KAESER P S, PIHAN G, CHURCH G M, YIN P. Nat. Biotechnol. , 2019, 37(9): 1080-1090.
[46] KISHI J Y, LAPAN S W, BELIVEAU B J, WEST E R, ZHU A, SASAKI H M, SAKA S K, WANG Y, CEPKO C L, YIN P.Nat. Methods, 2019, 16(6): 533-544.
[47] MERRITT C R, ONG G T, CHURCH S E, BARKER K, DANAHER P, GEISS G, HOANG M, JUNG J, LIANG Y, MCKAYFLEISCH J, NGUYEN K, NORGAARD Z, SORG K, SPRAGUE I, WARREN C, WARREN S, WEBSTER P J, ZHOU Z,ZOLLINGER D R, DUNAWAY D L, MILLS G B, BEECHEM J M. Nat. Biotechnol. , 2020, 38(5): 586-599.
[48] FARREN M R, SAYEGH L, WARE M B, CHEN H R, GONG J, LIANG Y, KRASINSKAS A, MAITHEL S K, ZAIDI M,SARMIENTO J M, KOOBY D, PATEL P, EL-RAYES B, SHAIB W, LESINSKI G B. JCI Insight, 2020, 5(1): e130362.
[49] BENDALL S C, SIMONDS E F, QIU P, AMIR E D, KRUTZIK P O, FINCK R, BRUGGNER R V, MELAMED R, TREJO A, ORNATSKY O I, BALDERAS R S, PLEVRITIS S K, SACHS K, PE’ER D, TANNER S D, NOLAN G P. Science, 2011,332(6030): 687-696.
[50] KEREN L, BOSSE M, THOMPSON S, RISOM T, VIJAYARAGAVAN K, MCCAFFREY E, MARQUEZ D, ANGOSHTARIR, GREENWALD N F, FIENBERG H, WANG J, KAMBHAM N, KIRKWOOD D, NOLAN G, MONTINE T J, GALLI S J,WEST R, BENDALL S C, ANGELO M. Sci. Adv. , 2019, 5(10): eaax5851.
