秈粳雜種不育的遺傳分析和候選基因鑒定
2024-04-24 02:08:14許娜唐穎徐正進孫健徐銓
中國農業科學 2024年8期
許娜,唐穎,徐正進,孫健,徐銓
秈粳雜種不育的遺傳分析和候選基因鑒定
許娜,唐穎,徐正進,孫健,徐銓
沈陽農業大學水稻研究所,沈陽 110866
【目的】秈粳亞種間雜交F1的育性問題嚴重阻礙了亞種間雜種優勢的利用,探究秈粳雜種不育的遺傳機理,挖掘新的秈粳雜種不育調控基因,為促進秈粳雜種結實率遺傳改良提供理論依據。【方法】粳稻品種Sasanishiki和秈稻品種Habataki雜交后,采用單粒傳法自交10代,獲得包含95個株系的穩定遺傳重組自交系(RIL),基于Illumina平臺,對雙親和RIL進行高通量測序,在全基因組水平分析RIL中Habataki血緣的分布與比例,進而鑒定偏分離區域作為潛在的秈粳雜種不育位點。同時,剖析“全球3000份水稻核心種質資源重測序計劃”中典型秈粳稻基因組變異數據,對群體水平進一步驗證并趨近目標育性基因區域。最終通過序列比對鎖定秈粳雜種不育候選基因。應用CRISPR基因編輯技術進行定點基因敲除,對目標基因進行功能驗證。【結果】Habataki和Sasanishiki的雜交F1在穗數、每穗粒數和千粒重上體現出明顯的雜種優勢,但其結實率顯著降低,I2-KI鏡檢發現F1花粉育性顯著降低。RIL的全基因組高通量測序在第1、3、5、6、7和12染色體上檢測到明顯的偏分離,即該區域的基因型趨向于秈稻Habataki。通過序列比對,進一步確定已知育性基因、和為第3、6和12染色體上偏分離區域的目標基因。應用CRISPR基因編輯技術敲除Habataki中的多拷貝,成功改善了其與Sasanishiki雜交F1的花粉育性和結實率,說明該基因可獨立行使功能。同時,在第1染色體的偏分離區域發現Habataki和Sasanishiki之間存在復雜的結構性變異,Sasanishiki基因組中一段24.7 kb包含4個預測基因的片段在Habataki中被一段64.8 kb包含10個預測基因的片段取代,此結構性變異可能參與秈粳雜種育性的調控。【結論】檢測到多個秈粳雜種不育相關位點,應用CRISPR基因編輯技術敲除Habataki中的多拷貝成功改善其雜種F1育性,確定其為水稻亞種間育性改良的目標基因。同時鎖定了第1染色體區域的目標基因。
水稻;秈粳雜種不育;高通量測序;基因編輯;候選基因
0 引言
【研究意義】水稻秈粳亞種間雜種優勢突出,可作為提高水稻單產的重要途徑,但是秈粳間雜種育性較低,限制了秈粳雜種優勢的育種應用。因此,闡明水稻秈粳雜種不育的遺傳機制,挖掘新的秈粳雜種育性調控基因,深入解析育性調控的分子機理,將為秈粳雜交育種與雜種優勢利用取得新突破提供理論基礎和種質資源。【前人研究進展】水稻是世界重要糧食作物,為全球約一半人口提供主食,其中90%以上集中在亞洲。亞洲栽培稻分為秈(/)和粳(/)2個亞種,在長期自然選擇和人工選擇過程中,秈粳稻基因組不斷分化,生態適應性等生物學特性產生顯著差異,農藝性狀各有利弊[1-4]。秈稻廣泛分布在印度、中國、東南亞,約占10%的粳稻集中分布在中國、日本、韓國等[5]。中國是世界上唯一同時大面積種植秈稻和粳稻的國家,約占2/3的秈稻主要分布于緯度和海拔較低地區,粳稻主要分布于緯度和海拔較高地區,中部地區秈粳交錯。我國秈粳并重,促使相關遺傳基礎研究成為得天獨厚且成果卓著的優勢研究領域[6-8]。雜交水稻技術的開發和應用為水稻增產作出了巨大貢獻[9-10]。在過去的幾十年里,僅在中國,雜交水稻每年就種植約1 700萬hm2(約占水稻總種植面積的60%),糧食產量比自交系品種提高了約20%[10]。然而,目前雜交水稻品種主要由同一亞種的品系之間的雜交培育而成,大多數是秈稻品系,由于親本系的遺傳多樣性狹窄,雜交產量已達到平穩水平,導致雜種優勢較低。相比之下,粳稻和秈稻品種之間的雜交種具有更強的抗逆性,因此,具有進一步提高產量潛力的巨大前景。在秈粳稻雜交中,多個秈粳雜種不育基因座的累積遺傳效應導致花粉和小穗的受精率非常低,嚴重影響雜交種結實率,因此,秈粳雜種不育是阻礙利用秈粳間雜種優勢獲得更高產量的主要障礙,克服秈粳雜種不育仍然是高產水稻育種的主要挑戰。遺傳學研究已發現大約50個與雜種不育相關的基因位點,提出了2種解釋水稻雜種不育模型,單位點孢子-配子體相互作用模型和重復配子-致死模型[11-12]。前者指孢子體細胞中單個基因位點的不同等位基因之間的遺傳相互作用會導致攜帶特定等位基因的配子致死;后者指2個基因座的不同等位基因之間的上位相互作用導致雜種不育。迄今為止,已經成功克隆的秈粳雜種不育基因位點主要有[13]、[14]、[15]、[16]、/[17]和[18]。【本研究切入點】秈粳雜種不育是受多個基因控制,依靠某一種或幾種不育基因位點的等位基因相互作用很難完全解釋秈粳不育遺傳機理,更多秈粳亞種雜種不育位點的挖掘將深入育性調控的分子遺傳機理研究,尤其是發掘到可獨立提升雜種育性的關鍵基因位點,將為水稻秈粳雜種優勢利用作出貢獻。【擬解決的關鍵問題】本研究圍繞秈粳雜種不育遺傳分析和調控基因位點挖掘這一關鍵科學問題,以秈粳交重組自交系為試驗試材,應用高通量測序和CRISPR基因編輯等方法,探索秈粳雜種不育的遺傳機理,挖掘新的秈粳不育調控位點,為亞種間雜交育性的遺傳改良提供理論依據。
1 材料與方法
1.1 供試水稻材料
以粳稻品種笹錦Sasanishiki(Sasa)和秈稻品種Habataki(Haba)為雙親雜交獲得F1植株,采用單粒傳法套袋自交10代,獲得包含95個株系的穩定遺傳重組自交系(RIL)。親本、F1植株和重組自交系于2022年春季4月23日播種,5月23日種植于沈陽農業大學水稻所試驗田(41°N,123°E),每個株系按照3行×10株規模種成小區,株距為10 cm,行距為30 cm,每穴單苗插植,常規水肥管理。
1.2 表型鑒定
抽穗后45 d,每小區取中部5株收獲,充分曬干后,考察單株穗數。取長勢均勻的5穗,統計每穗粒數和結實率。隨機取100粒飽滿籽粒稱重統計千粒重。最后運用Microsoft Excel 2021計算各材料每株的各性狀平均值、標準差,并對各性狀進行兩尾等方差檢驗,使用GraphPad Prism 8進行作圖。用1% I2-KI溶液進行花粉育性的鑒定。
1.3 DNA提取與全基因組測序和遺傳圖譜構建
插秧后3周,取幼嫩葉片,采用CTAB法提取DNA,送北京百邁克生物科技有限公司進行高通量測序分析,重組自交系的測序共獲得207.50 Gb數據,平均深度為5.46×。使用Lep-MAP3(ref)軟件的OrderMarkers2模塊,計算標記間的遺傳距離和各個染色體的圖距,主要參數設置為:identical Limit=0.01,min Error=1e-5,sex Averaged=0,informative Mask=123,use Kosambi=1,其他參數使用默認值。利用劃bin策略進行圖譜構建,得到2 241個bins。
1.4 RNA提取和qRT-PCR
使用TRIzol試劑(Invitrogen,USA)從Sasa、Haba及其雜交種的水稻組織(花藥、幼穗、葉、莖和根)中提取總RNA。RNA樣品用DNaseⅠ(Promega,USA)處理。使用Superscript Ⅲ逆轉錄酶(Invitrogen,USA)和寡聚dT或-特異性引物從2—3 μg總RNA合成第一鏈cDNA。以1()為內參,對進行qRT-PCR分析,試驗設置3次生物學重復,并用雙尾Student’s檢驗分析其顯著性。SYBR Green QPCR混合物(Bio-Rad)用于qRT-PCR。使用Bio-rad CFX Connect實時PCR系統進行反應:95 ℃ 3 min;95 ℃ 10 s,60 ℃15 s,72 ℃ 20 s,42個循環。()在Sasa中的表達模式參考自水稻表達譜數據庫(http://ricexpro.dna. affrc.go.jp)。
1.5 CRISPR/Cas9基因編輯
以秈稻品種Haba為遺傳背景材料,進行重組載體轉化。轉化用的農桿菌為EHA105菌種,原核感受態細胞為大腸桿菌DH5α菌株。根據和序列存在多態性的區間,運用華南農業大學亞熱帶農業生物資源保護與利用國家重點實驗室劉耀光院士團隊開發的基因編輯工具包CRISPR-GE(http://skl.scau.edu.cn)進行靶位點設計,基因編輯靶點序列、引物合成及測序服務均由華大基因完成。參照Li等[19]方法進行基因編輯載體的構建和基因編輯植株的遺傳轉化和篩選。
1.6 秈粳亞種群體基因組分化分析
基于“全球3000份水稻核心種質資源重測序計劃”(3K Rice Project)的數據庫中樣本背景信息,首先根據群體結構Structure信息去掉中間型組群-adm、-adm、aus(普遍具有廣親和性)、bus、admix。在剩下的秈稻亞種-1a、-1b、-2、-3中,將Structure模式圖中體現為-2與aus血緣互滲的種質材料刪掉,最后保留209份-1a、205份-1b、475份-3,粳稻亞種中保留319份-tmp與388份-trp的材料。由于現代水稻育種已將亞種間生殖隔離打破,目前主栽品種中很多具有混合的亞種血緣,因此,在篩選到的秈粳種質中,進一步選擇農家品種將使其血統更加純粹。最終在3K Rice Project數據庫中篩選并下載424份典型秈稻農家品種(landrace-I)與238份典型粳稻農家品種(landrace-J)的相關數據。應用bcftools軟件提取上述2個亞種入選種質的vcf文件。應用vcftools軟件分別計算全部染色體、第1染色體的100 kb窗口與第1染色體目標QTL區域單獨SNP的群體分化系數st,利用顯著峰值判定在群體中基因組分歧強烈的SNP位點,作為生殖隔離的基因組響應位點,可視化用R的CMplot包。
2 結果
2.1 F1雜種優勢
調查粳稻品種Sasa、秈稻品種Haba及其雜交F1,發現秈粳雜交F1展現出雜種優勢(圖1-A—B)。雙親株高沒有顯著差別,F1代株高顯著高于雙親(圖1-C)。Sasa穗數顯著多于Haba,雜交F1顯著多于Sasa(圖1-D)。Haba每穗粒數顯著多于Sasa,雜交F1每穗粒數超過200,顯著多于Haba(圖1-E)。Sasa和Haba之間千粒重沒有顯著差異,雜交F1千粒重顯著高于雙親(圖1-F)。Sasa和Haba之間結實率沒有顯著差異,達到90%,雜交F1結實率顯著降低,僅達到20%(圖1-G),不育籽粒并未受精,穎殼內空癟,不育籽粒均勻分布在整個穗部。因此,F1籽粒結實率降低是限制發揮秈粳間雜種優勢利用的主要問題。

A:株型;B:穗型;C:株高;D:穗數;E:每穗粒數;F:千粒重;G:結實率。不同字母表示差異顯著(P<0.05)。下同
2.2 F1的花粉育性
Sasa和Haba雜交F1結實率顯著降低,使用I2-KI鏡檢進行雙親和雜交F1花粉的育性檢測。結果顯示,Sasa和Haba的花粉表現出完全可育,可染色花粉比例沒有顯著差別,均達到90%(圖2-A—B和圖2-D),而雜交F1的可染色花粉比例顯著降低,不足40%。不育花粉體積較小,呈圓球形或不規則形,空癟無物,著色淺或不著色,大多為典型的空敗花粉,少量為圓敗花粉(圖2-C—D)。因此,推測花粉育性降低是秈粳雜種F1結實率降低的原因之一。因為雜交F1結實率為20%,顯著低于其花粉可染色比例,推測雜交F1存在一定比例的雌配子不育(圖1-G和圖2-C—D)。
2.3 F1雜種不育的遺傳分析
為了闡明調控F1雜種不育的基因位點,對Sasa和Haba雜交后自交10代構建的包含95個株系的重組自交系(RIL)進行基于Illumina平臺的全基因組高通量測序。重組自交系的測序共獲得207.50 Gb數據,平均深度為5.46×,Haba測序得到22.94 Gb數據,測序深度為51.00×,Sasa測序得到23.24 Gb數據,測序深度為56.00×。比對雙親鑒定到1 947 668個SNP,過濾后得到1 591 495個高質量SNP進行遺傳圖譜構建,得到一個包含2 241個bins的遺傳圖譜(圖3-A—B)。根據RIL的基因型,分析其Haba類型SNP在群體中的比例,繪制秈型血緣滲入比率示意圖,發現雖然使用單粒傳法構建RIL,但Haba血緣平均比例在全基因組中并不是維持在50%左右,其平均值為0.38,其中,第4、9、11和12染色體Haba血緣比例較低,第7染色體Haba血緣整體較高。在第1、3、6、7和12染色體檢測到明顯的偏分離,即該區域的基因型趨向于秈稻Haba,Haba血緣超過了平均值的2倍,超過了0.76,說明該區域在雜交后分離過程中有更多的秈稻Haba型配子保留下來(圖3-C),暗示這些區域可能存在調控秈粳雜種育性的關鍵基因。

A:Sasa花粉I2-KI鏡檢;B:Haba花粉I2-KI鏡檢;C:F1(Sasa/Haba)花粉I2-KI鏡檢;D:Sasa、Haba和F1(Sasa/Haba)花粉可染色率
2.4 秈粳雜種不育基因序列比對
比對前人關于秈粳雜種不育調控位點的研究后發現第3、6和12染色體的偏分離峰與已克隆的關鍵秈粳雜種不育基因、和位置重合[14-16]。比對Sasa和Haba的位點的3個緊密相連的連鎖基因開放閱讀框(、和),發現Sasa和Haba為典型的粳稻和秈稻位點(圖4);對比Sasa和Haba的位點,發現Sasa在位點只包含一個拷貝,但是Haba在該位點包含3個拷貝、和(圖4)。比對(和)位點,發現位點在雙親之間存在豐富的多態性,并且位點的T缺失和2個SNP,導致移碼突變并在外顯子形成終止密碼子。在本群體中并沒有檢測到已經克隆的秈粳雜種不育相關基因/、和[13, 17-18]。進一步比對這些基因在Sasa和Haba之間的多態性,結果顯示,在Haba的位點并不存在與Kasalath相同的518 bp的轉座子插入;比對(和)位點,發現Haba的位點序列與Sasa完全一致,這可能是本群體中沒有檢測到/和位點的原因。經比對發現Sasa和Haba在位點存在結構性變異,其中Sasa中一段11.2 kb包含和的片段被一段35.0 kb包含5個ORF(、、/、/和)的片段取代,但是在本群體中并沒有檢測到該位點的偏分離現象,暗示在Sasa和Haba的雜合背景下,的功能被其他育性恢復位點的遺傳效應所補償。
2.5 秈粳雜種不育位點Sc的基因編輯驗證
Haba在位點存在多拷貝現象,其中,在第一個外顯子中有一段2.0 kb的插入,并且缺失了第一個外顯子的末端,導致其編碼的DUF1618功能域缺失,此外,還在第一個內含子中有一段10.1 kb的大片段插入,因此,認為并不具備位點的功能,是一個無功能基因(圖4-C)。秈稻Haba中的另外2個拷貝和均含有完整的拷貝,與的第一個外顯子序列相似度分別為97.5%和97.8%,第二個外顯子與完全一致(圖5-A)。Sasa的單拷貝僅在花藥中表達,而Haba的多拷貝使其在花藥中表達量顯著增加,而且在根、莖、葉和穗中均有較高表達(圖5-B)。

A:重組自交系的遺傳圖譜;B:重組自交系中Haba血緣的滲入比例;C:已知秈粳雜種不育位點

A:DPL1和DPL2位點的序列比對;B:SaF和SaM位點的序列比對;C:Sc位點的結構性變異;D:S5位點的序列比對;E:RHS12位點的結構性變異;F:HSA1a和HSA1b位點的序列比對
雖然和序列相似,但其在第一個外顯子中仍然存在細微差別,其中,一個G堿基的插入形成了中不存在的CRISPR基因編輯的靶點PAM序列CGG。以此PAM序列設計了一個只敲除拷貝,但是無法敲除拷貝的CRISPR載體,對Haba進行基因編輯。在T2代篩選到2個Cas free的純合突變CR-1和CR-2,分別缺失了1個C堿基和5個堿基AGGCC,2個突變類型均引起移碼突變,缺失了DUF1618功能域(圖6-A)。對比CR-1和CR-2與Sasa的雜交F1植株和Sasa與Haba的雜交F1植株發現F1(Sasa/CR-1)和F1(Sasa/CR-2)的花粉可染色比例顯著提高(圖6-B)。表達量分析發現F1(Sasa/Haba)的表達量顯著降低,暗示和會抑制的表達,但是F1(Sasa/CR-1)和F1(Sasa/ CR-2)的表達量顯著高于F1(Sasa/Haba)的表達量(圖6-C),暗示減少的拷貝數可以減弱其對的抑制。I2-KI鏡檢發現F1(Sasa/CR-1)和F1(Sasa/CR-2)的花粉育性顯著高于F1(Sasa/Haba)(圖6-D),并且其結實率也顯著高于F1(Sasa/Haba)(圖6-E)。
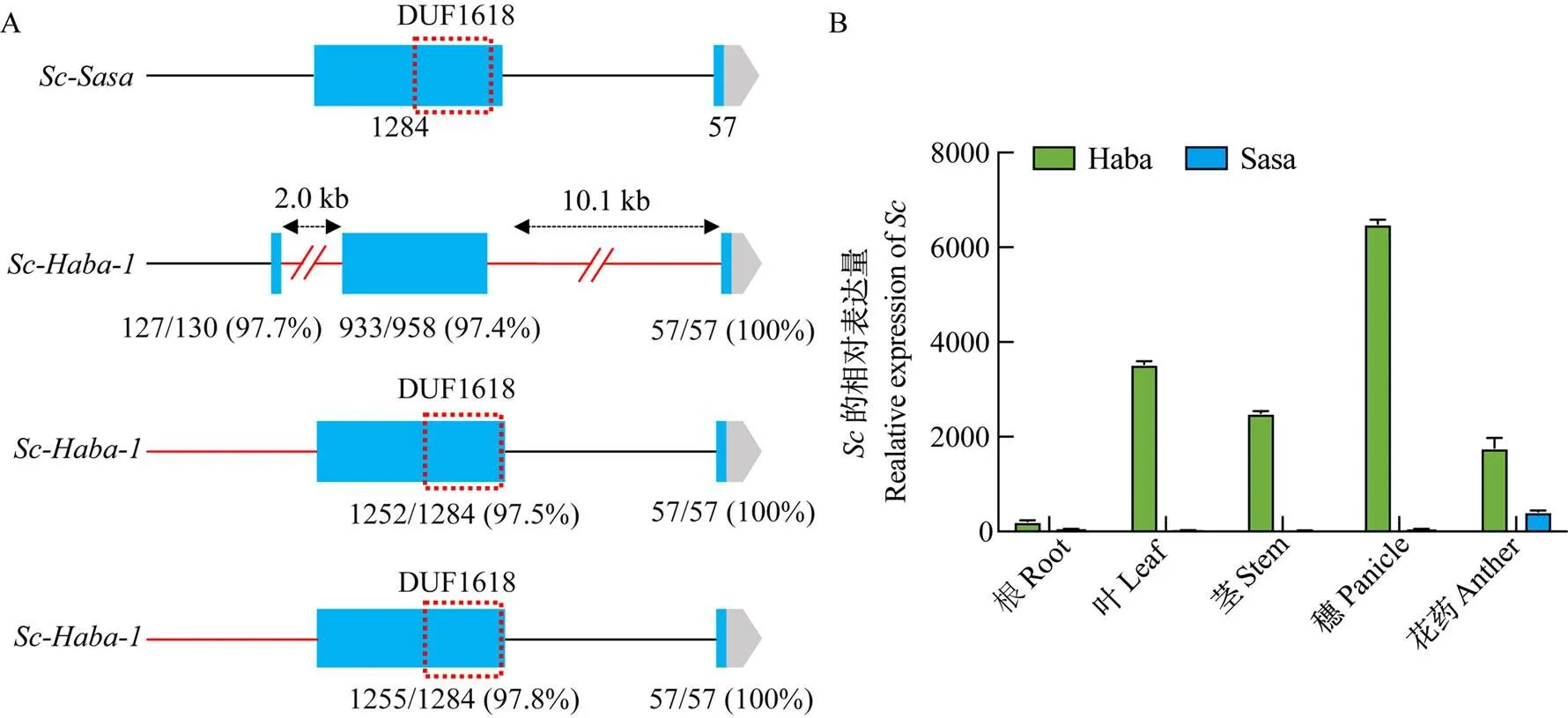
A:Haba中Sc位點的多拷貝;B:Sc在Sasa和Haba中的表達模式
2.6 秈粳雜種不育位點Sd的候選基因預測
進一步分析第1染色體3.1 Mb附近區域內的偏分離信號,該信號峰的區域作為亞種間雜交不育位點在臺中65的近等基因系中被報道,然而,目標基因并沒被成功地鑒定與克隆[20-21]。為了在群體水平中驗證該位點是否具有亞種間生殖隔離的普遍性,本研究剖析了“全球3000份水稻核心種質資源重測序計劃”中典型秈粳稻基因組變異數據。應用vcftools軟件分析了入選典型秈粳稻群體間在第1染色體上每100 kb的分化系數st,發現亞種間st分化信號峰值出現在3.8 Mb物理位置,并輻射上下游1 Mb基因組區域,與RIL群體偏分離信號重疊(圖7)。在群體水平上證實了該位點是亞種間生殖隔離的普遍性關鍵位點。因此,比對該基因組區域內Sasa和Haba的預測基因的序列差別,發現Sasa和Haba在該區間內存在復雜的基因組結構性變異,Sasa基因組中一段24.7 kb的片段在Haba中被一段64.8 kb的片段取代。Sasa基因組中24.7 kb的片段中包含、、和4個預測基因,而Haba的第1染色體64.8 kb片段中包含10個預測基因,且Sasa中的4個預測基因在Haba中無法比對到相似序列(圖7)。綜上所述,粳稻Sasa該區域內的4個預測基因和秈稻Haba該區域內的10個預測基因可能在其雜種育性調控上起重要作用。
3 討論
3.1 秈粳雜種不育的遺傳機理
根據配子敗育的類型不同,秈粳雜種不育分為雌配子敗育和雄配子敗育,其中,利用臺中65的近等基因系,對秈粳雜交花粉敗育進行系統的遺傳研究,先后定位了、、、和等5個秈粳雜種花粉不育位點[20-23],其中、和位點已經被成功克隆[13-14, 24]。為一個由2個相鄰基因和組成的雜種不育復合基因位點,這兩個基因分別編碼一個F-盒蛋白和一個小的泛素樣修飾物E3連接酶。在雜合子中,來自3個等位基因(秈型和,以及粳稻)的蛋白質之間的有害相互作用導致攜帶粳稻等位基因的花粉粒選擇性致死,從而在分子水平上建立了雙基因/三成分相互作用模型[13]。位點秈粳等位基因間的DNA序列存在復雜的結構變異,秈稻等位基因攜帶2—3個的同源拷貝,因此,秈粳雜種中的高表達抑制了花粉中的表達,進而造成攜帶的花粉敗育[14]。近期研究發現華南農業大學劉自強團隊克隆的位點[24]與南京農業大學萬建民院士團隊克隆的秈粳雜種不育基因[18]及華中農業大學歐陽亦聃教授團隊報道的[25]為同一位點,該位點由緊密連鎖的2個基因組成,可以分別比喻為“破壞者”和“守衛者”。“破壞者”對所有花粉產生傷害作用,引起花粉的敗育;而“守衛者”阻止“破壞者”的傷害作用,因此,那些遺傳了該基因的花粉,因受到保護能正常發育。和被定位到第1染色體和第5染色體上,目前還沒有成功克隆[20, 22-23]。本研究在第1染色體上鑒定到的偏分離峰與定位區間重合,并且在“全球3000份水稻核心種質資源重測序計劃”的群體水平上證實該位點是一個亞種間生殖隔離的普遍性位點。通過序列比對發現粳稻Sasa基因組中一段24.7 kb的片段在Haba中被一段64.8 kb的片段取代,進而提名了候選基因,為該基因的克隆與功能研究提供了關鍵線索。秈粳分化位點的分析也佐證了該區域為秈粳分化位點,暗示該區域可能調控秈粳雜種不育。/是一對同源基因,均編碼植物特異小分子蛋白,對花粉萌發有重要作用。典型秈稻攜帶有功能的和無功能的,典型粳稻攜帶有功能的和無功能的,而秈粳雜種中攜帶2個功能缺失突變等位基因的花粉,不能萌發,這意味著在花粉萌發中扮演著一個重要的角色[17],本研究發現Haba并不含有典型的秈型等位基因。是一個復雜的基因座,調控雌配子敗育,包含3個緊密連接的基因、和。在雌性孢子發生過程中,秈稻等位基因的殺手/伴侶基因和的共同作用導致內質網應激和胚胎囊的流產,其中,粳稻等位基因中含有非功能保護基因;秈稻等位基因的功能性拯救了這些效應[15]。基因座包含2個緊密連接的基因,和;粳稻和秈稻雜交種中基因的粳稻和秈稻等位基因之間的上位性相互作用導致雌配子致死[16]。本研究在第5染色體和第12染色體鑒定到的偏分離峰與和位置重合,佐證了和所引起的雌配子敗育也是引起Sasa和Haba雜種結實率下降的原因。
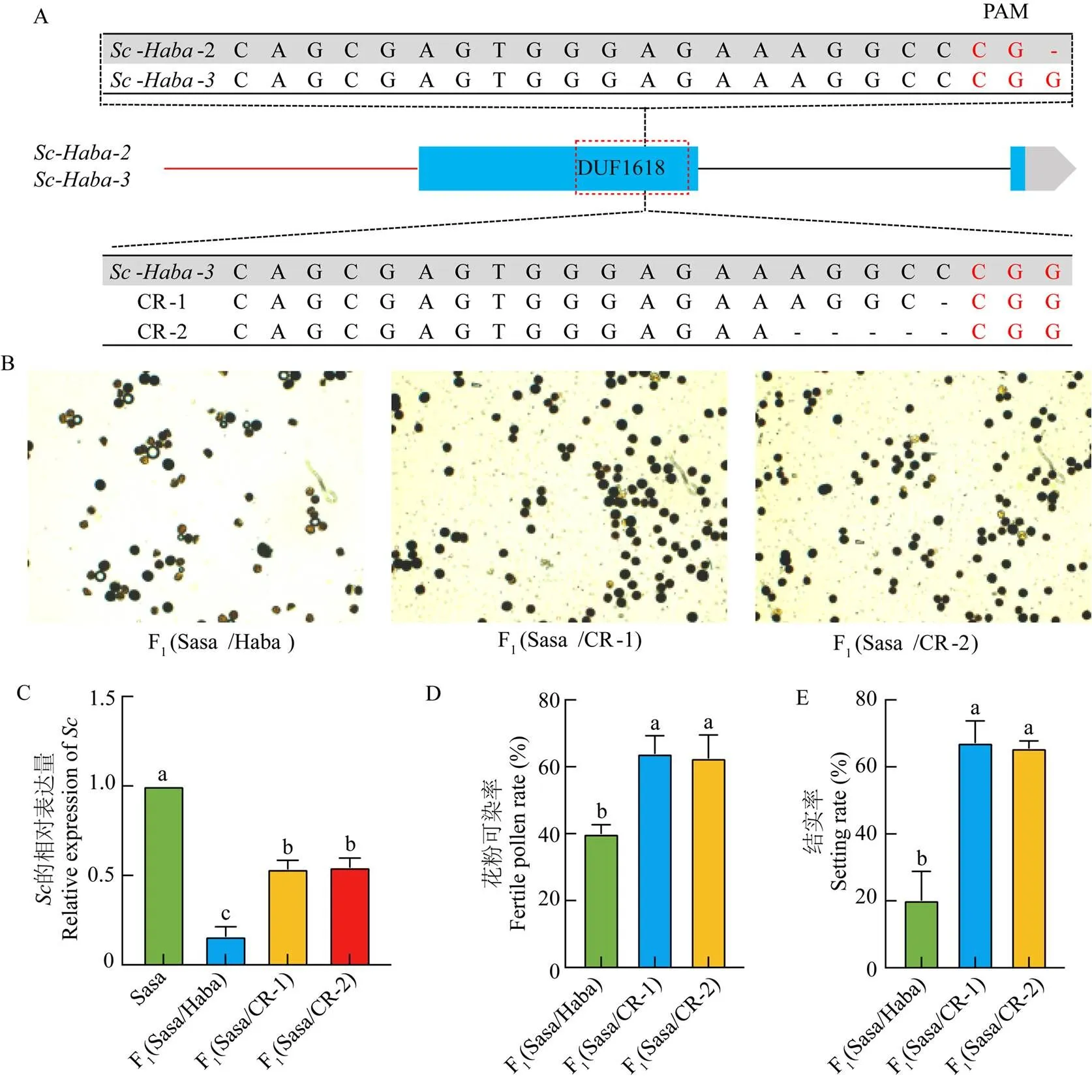
A:Sc-Haba-2與Sc-Haba-3靶點PAM序列的差異和基因編輯植株的突變序列;B:F1(Sasa/Haba)、F1(Sasa/CR-1)和F1(Sasa/CR-2)植株的花粉I2-KI鏡檢;C:Sasa、F1(Sasa/Haba)、F1(Sasa/CR-1)和F1(Sasa/CR-2)中Sc-Sasa的表達量;D:F1(Sasa/Haba)、F1(Sasa/CR-1)和F1(Sasa/CR-2)的花粉可染率;E:F1(Sasa/Haba)、F1(Sasa/CR-1)和F1(Sasa/CR-2)的結實率

A:Chr.1的100 kb窗口10 kb滑動的群體分化Fst區域,粉色區域表示3.8 Mb物理位置處Fst分化信號峰值所輻射的上下游1 Mb基因組區域;B:第1染色體中Haba基因型的滲入比例;C:候選區間內秈粳間的基因組結構性變異
3.2 結構性變異影響秈粳雜種育性
基因組結構變異,包括片段插入、缺失、反轉、易位、重復和拷貝數變異,對各種生物過程、農藝性狀和人類疾病具有顯著的遺傳影響[26]。在三代長讀長測序技術推廣以前,基因組結構性變異,尤其是基因拷貝數變異,很難通過圖位克隆鑒定。拷貝數變異在植物基因組中廣泛分布[27]。最近報道的泛基因組揭示了隱藏的拷貝數變異,并證明拷貝數變異調節重要的農藝性狀[25, 28-29]。基因座的拷貝數變化有助于水稻的粒型多樣性[29],基因座的額外拷貝數顯著增加了每穗的粒數[30-31],的雙拷貝可能是解釋Koshihikari早花表型的原因候選者[26, 32]。基因拷貝數變異也參與水稻秈粳雜種育性的調控,的拷貝數變異在秈粳亞種之間分化,秈稻一般含有2—3個拷貝,而粳稻中只有一個拷貝[14]。利用RGP網站的RiceGAAS系統對座位定位區段進行預測,發現秈稻每個拷貝中還含有預測基因、、和,并且、、均為位點雜種不育功能所必需[33]。本研究還在和拷貝中發現了、、和預測基因,暗示位點可能存在超過10個預測基因參與的復雜遺傳機理。//位點也存在秈粳間復雜的結構性變異,序列分析顯示,在臺中65中,定位區包含2個ORF(命名為和),而在秈稻G4中,除了與或同源的基因(分別命名為和)外,它還包含3個額外的ORF,命名為、和,而這額外的基因對秈粳雜種育性有重要的調控作用[18, 24-25]。本研究定位的位點區間同樣在秈粳之間存在復雜的結構性變異,粳稻Sasa該區域含有4個預測基因,而秈稻Haba該區域含有10個預測基因,暗示這些ORF中可能存在調控秈粳雜交育性的關鍵基因。
3.3 秈粳雜種優勢利用
作物雜種優勢利用是大幅提高糧食產量的重要途徑,一般來說品種間親緣關系越遠,雜交優勢越明顯。秈稻和粳稻亞種間能育成超級雜交稻可以比現有雜交水稻增產15%以上[34]。然而秈稻和粳稻之間存在嚴重的生殖隔離,是阻礙雜種優勢利用的最大障礙之一。深入研究秈粳雜種不育遺傳機理將為秈粳雜種優勢的育種應用提供理論依據和種質資源。隨著現代分子生物育種技術的發展,越來越多的雜種不育座位被鑒定、克隆,為秈粳稻雜種育性改良提供了潛在的靶點。應用CRISPR基因編輯技術可以在已知的雜交不育基因位點進行點突變或者大片段敲除,可改良秈粳雜種育性,從而加快廣親和系的培育,如,通過敲除座位的拷貝中的1個或2個,減少的基因表達量,從而恢復的正常表達,創制出等位基因[14];應用 CRISPR方法對基因座2個基因和進行編輯,快速創建等位基因也成功改良了秈粳雜種育性[35]。本研究也通過CRISPR基因編輯,應用的特異PAM序列將其敲除,改善了其與Sasa的雜種育性。重要的是該基因可獨立行使功能來改善其雜種F1育性,可作為水稻亞種間育性改良的目標基因,具有較大的應用潛力。
4 結論
在第1、3、6、7和12染色體檢測到可能存在調控秈粳雜種育性關鍵基因的偏分離區域。應用CRISPR基因編輯技術敲除Haba第3染色體上位點的多拷貝成功改善其秈粳雜種F1育性。同時發現第1染色體上位點秈粳間復雜結構性變異是調控亞種間生殖隔離的關鍵,并提名了目標候選基因。
[1] GARRIS A J, TAI T H, COBURN J, KRESOVICH S, McCOUCH S. Genetic structure and diversity inL.. Genetics, 2005, 169(3): 1631-1638.
[2] HUANG X H, WEI X H, SANG T, ZHAO Q, FENG Q, ZHAO Y, LI C Y, ZHU C R, LU T T, ZHANG Z W, LI M, FAN D L, GUO Y L, WANG A H, WANG L, DENG L W, LI W J, LU Y Q, WENG Q J, LIU K Y, HUANG T, ZHOU T Y, JING Y F, LI W, LIN Z, BUCKLER E S, QIAN Q, ZHANG Q F, LI J Y, HAN B. Genome-wide association studies of 14 agronomic traits in rice landraces. Nature Genetics, 2010, 42(11): 961-967.
[3] HUANG X H, ZHAO Y, WEI X H, LI C Y, WANG A H, ZHAO Q, LI W J, GUO Y L, DENG L W, ZHU C R, FAN D L, LU Y Q, WENG Q J, LIU K Y, ZHOU T Y, JING Y F, SI L Z, DONG G J, HUANG T, LU T T, FENG Q, QIAN Q, LI J Y, HAN B. Genome-wide association study of flowering time and grain yield traits in a worldwide collection of rice germplasm. Nature Genetics, 2012, 44(1): 32-39.
[4] YU H, LIN T, MENG X B, DU H L, ZHANG J K, LIU G F, CHEN M J, JING Y H, KOU L Q, LI X X, GAO Q, LIANG Y, LIU X D, FAN Z L, LIANG Y T, CHENG Z K, CHEN M S, TIAN Z X, WANG Y H, CHU C C, ZUO J R, WAN J M, QIAN Q, HAN B, ZUCCOLO A, WING R A, GAO C X, LIANG C Z, LI J Y. A route to de novo domestication of wild allotetraploid rice. Cell, 2021, 184(5): 1156-1170.
[5] WANG W S, MAULEON R, HU Z Q, CHEBOTAROV D, TAI S S, WU Z C, LI M, ZHENG T Q, FUENTES R R, ZHANG F, MANSUETO L, COPETTI D, SANCIANGCO M, PALIS K C, XU J L, SUN C, FU B Y, ZHANG H L, GAO Y M, ZHAO X Q, SHEN F, CUI X, YU H, LI Z C, CHEN M L, DETRAS J, ZHOU Y L, ZHANG X Y, ZHAO Y, KUDRNA D, WANG C C, LI R, JIA B, LU J Y, HE X C, DONG Z T, XU J B, LI Y H, WANG M, SHI J X, LI J, ZHANG D B, LEE S, HU W S, POLIAKOV A, DUBCHAK I, ULAT V J, BORJA F N, MENDOZA J R, ALI J, LI J, GAO Q, NIU Y C, YUE Z, NAREDO M E B, TALAG J, WANG X Q, LI J J, FANG X D, YIN Y, GLASZMANN J C, ZHANG J W, LI J Y, HAMILTON R S, WING R A, RUAN J, ZHANG G Y, WEI C C, ALEXANDROV N, MCNALLY K L, LI Z K, LEUNG H. Genomic variation in 3,010 diverse accessions of Asian cultivated rice. Nature, 2018, 557(7703): 43-49.
[6] WEI X, QIU J, YONG K C, FAN J J, ZHANG Q, HUA H, LIU J, WANG Q, OLSEN K M, HAN B, HUANG X H. A quantitative genomics map of rice provides genetic insights and guides breeding. Nature Genetics, 2021, 53(2): 243-253.
[7] LIU Y Q, WANG H R, JIANG Z M, WANG W, XU R N, WANG Q H, ZHANG Z H, LI A F, LIANG Y, OU S J, LIU X J, CAO S Y, TONG H N, WANG Y H, ZHOU F, LIAO H, HU B, CHU C C. Genomic basis of geographical adaptation to soil nitrogen in rice. Nature, 2021, 590(7847): 600-605.
[8] ZHOU G, CHEN Y, YAO W, ZHANG C J, XIE W B, HUA J P, XING Y Z, XIAO J H, ZHANG Q F. Genetic composition of yield heterosis in an elite rice hybrid. Proceedings of the National Academy of Sciences of the United States of America, 2012, 109(39): 15847-15852.
[9] 袁隆平. 雜交水稻的育種戰略設想. 雜交水稻, 1987(1): 1-3.
YUAN L P. The strategy of hybrid rice breeding. Hybrid Rice,1987(1): 1-3. (in Chinese)
[10] CHENG S H, ZHUANG J Y, FAN Y Y, DU J H, CAO L Y. Progress in research and development on hybrid rice: a super-domesticate in China. Annals of Botany, 2007, 100(5): 959-966.
[11] OKA H. Analysis of genes controlling f(1) sterility in rice by the use of isogenic lines. Genetics, 1974, 77(3): 521-534.
[12] SANO Y. The genic nature of gamete eliminator in rice. Genetics, 1990, 125(1): 183-191.
[13] LONG Y M, ZHAO L F, NIU B X, SU J, WU H, CHEN Y L, ZHANG Q Y, GUO J X, ZHUANG C X, MEI M T, XIA J X, WANG L, WU H B, LIU Y G. Hybrid male sterility in rice controlled by interaction between divergent alleles of two adjacent genes. Proceedings of the National Academy of Sciences of the United States of America, 2008, 105(48): 18871-18876.
[14] SHEN R X, WANG L, LIU X P, WU J, JIN W W, ZHAO X C, XIE X R, ZHU Q L, TANG H W, LI Q, CHEN L T, LIU Y G. Genomic structural variation-mediated allelic suppression causes hybrid male sterility in rice. Nature Communications, 2017, 8: 1310.
[15] YANG J Y, ZHAO X B, CHENG K, DU H Y, OUYANG Y D, CHEN J J, QIU S Q, HUANG J Y, JIANG Y H, JIANG L W, DING J H, WANG J, XU C G, LI X H, ZHANG Q F. A killer-protector system regulates both hybrid sterility and segregation distortion in rice. Science, 2012, 337(6100): 1336-1340.
[16] KUBO T, TAKASHI T, ASHIKARI M, YOSHIMURA A, KURATA N. Two tightly linked genes at thelocus cause both F1and F2hybrid sterility in rice. Molecular Plant, 2016, 9(2): 221-232.
[17] MIZUTA Y, HARUSHIMA Y, KURATA N. Rice pollen hybrid incompatibility caused by reciprocal gene loss of duplicated genes. Proceedings of the National Academy of Sciences of the United States of America, 2010, 107(47): 20417-20422.
[18] WANG C L, WANG J, LU J Y, XIONG Y H, ZHAO Z G, YU X W, ZHENG X M, LI J, LIN Q B, REN Y L, HU Y, HE X D, LI C, ZENG Y L, MIAO R, GUO M L, ZHANG B S, ZHU Y, ZHANG Y H, TANG W J, WANG Y L, HAO B Y, WANG Q M, CHENG S Q, HE X J, YAO B W, GAO J W, ZHU X F, YU H, WANG Y, SUN Y, ZHOU C L, DONG H, MA X D, GUO X P, LIU X, TIAN Y L, LIU S J, WANG C M, CHENG Z J, JIANG L, ZHOU J W, GUO H S, JIANG L W, TAO D Y, CHAI J J, ZHANG W, WANG H Y, WU C Y, WAN J M. A natural gene drive system confers reproductive isolation in rice. Cell, 2023, 186(17): 3577-3592.
[19] LI X K, WU L, WANG J H, SUN J, XIA X H, GENG X, WANG X H, XU Z J, XU Q. Genome sequencing of rice subspecies and genetic analysis of recombinant lines reveals regional yield- and quality- associated loci. BMC biology, 2018, 16(1): 102.
[20] LI W T, ZENG R Z, ZHANG Z M, DING X H, ZHANG G Q. Identification and fine mapping of, a new locus conferring the partial pollen sterility of intersubspecific F1hybrids in rice (L.). Theoretical and Applied Genetics, 2008, 116(7): 915-922.
[21] 張桂權, 盧永根, 張華, 楊進昌, 劉桂富. 栽培稻()雜種不育性的遺傳研究: Ⅳ.F1花粉不育性的基因型. 遺傳學報, 1994, 21(1): 34-41.
ZHANG G Q, LU Y G, ZHANG H, YANG J C, LIU G F. Genetic studies on the hybrid sterility in cultivated rice (): IV. Genotypes for F1pollen sterility. Acta Genetica Sinica, 1994, 21(1): 34-41. (in Chinese)
[22] LI W T, ZENG R Z, ZHANG Z M, DING X H, ZHANG G Q. Fine mapping of locusfor F1pollen sterility in rice (L.). Chinese Science Bulletin, 2006, 51(6): 675-680.
[23] ZHANG G Q. Prospects of utilization of inter-subspecific heterosis betweenandrice. Journal of Integrative Agriculture, 2020, 19(1): 1-10.
[24] WANG D Q, WANG H R, XU X M, WANG M, WANG Y H, CHEN H, PING F, ZHONG H H, MU Z K, XIE W T, LI X Y, FENG J B, ZHANG M L, FAN Z L, YANG T F, ZHAO J L, LIU B, RUAN Y, ZHANG G Q, LIU C L, LIU Z Q. Two complementary genes in a presence-absence variation contribute toreproductive isolation in rice. Nature Communications, 2023, 14(1): 4531.
[25] ZHOU P H, WANG Z J, ZHU X C, TANG Y, YE L, YU H H, LI Y T, ZHANG N K, LIU T, WANG T, WU Y Y, CAO D Y, CHEN Y, LI X, ZHANG Q L, XIAO J H, YU S B, ZHANG Q F, MI J M, OUYANG Y D. A minimal genome design to maximally guarantee fertile inter-subspecific hybrid rice. Molecular Plant, 2023, 16(4): 726-738.
[26] QIN P, LU H W, DU H L, WANG H, CHEN W L, CHEN Z, HE Q, OU S J, ZHANG H Y, LI X Z, LI X X, LI Y, LIAO Y, GAO Q, TU B, YUAN H, MA B T, WANG Y P, QIAN Y W, FAN S J, LI W T, WANG J, HE M, YIN J J, LI T, JIANG N, CHEN X W, LIANG C Z, LI S G. Pan-genome analysis of 33 genetically diverse rice accessions reveals hidden genomic variations. Cell, 2021, 184(13): 3542-3558.
[27] LYE Z N, PURUGGANAN M D. Copy number variation in domestication. Trends in Plant Science, 2019, 24(4): 352-365.
[28] SHANG L G, LI X X, HE H Y, YUAN Q L, SONG Y N, WEI Z R, LIN H, HU M, ZHAO F L, ZHANG C, LI Y H, GAO H S, WANG T Y, LIU X P, ZHANG H, ZHANG Y, CAO S M, YU X M, ZHANG B T, ZHANG Y, TAN Y Q, QIN M, AI C, YANG Y X, ZHANG B, HU Z Q, WANG H R, LV Y, WANG Y X, MA J, WANG Q, LU H W, WU Z, LIU S L, SUN Z Y, ZHANG H L, GUO L B, LI Z C, ZHOU Y F, LI J Y, ZHU Z F, XIONG G S, RUAN J, QIAN Q. A super pan-genomic landscape of rice. Cell Research, 2022, 32(10): 878-896.
[29] WANG Y, LI F C, ZHANG F, WU L A, XU N, SUN Q, CHEN H, YU Z W, LU J H, JIANG K, WANG X C, WEN S Y, ZHOU Y, ZHAO H, JIANG Q, WANG J H, JIA R Z, SUN J, TANG L, XU H, HU W, XU Z J, CHEN W F, GUO A P, XU Q. Time-ordering/genomes analysis indicates the importance of large structural variants in rice breeding. Plant Biotechnology Journal, 2023, 21(1): 202-218.
[30] WANG Y X, XIONG G S, HU J, JIANG L, YU H, XU J, FANG Y X, ZENG L J, XU E B, XU J, YE W J, MENG X B, LIU R F, CHEN H Q, JING Y H, WANG Y H, ZHU X D, LI J Y, QIAN Q. Copy number variation at thelocus contributes to grain size diversity in rice. Nature Genetics, 2015, 47(8): 944-948.
[31] WU Y, WANG Y, MI X F, SHAN J X, LI X M, XU J L, LIN H X. The QTLencodes, which increases grain number and yield by increasing cytokinin activity in rice panicle meristems. PLoS Genetics, 2016, 12(10): e1006386.
[32] FORNARA F, PARENICOVá L, FALASCA G, PELUCCHI N, MASIERO S, CIANNAMEA S, LOPEZ-DEE Z, ALTAMURA M M, COLOMBO L, KATER M M. Functional characterization of, a member of thesubfamily ofbox genes. Plant Physiology, 2004, 135(4): 2207-2219.
[33] 吳疆. 水稻座位蛋白復合體參與秈粳雜種雄性不育的研究[D]. 廣州: 華南農業大學, 2017.
WU J. A protein complex of thelocus controlshybrid male sterility in rice[D]. Guangzhou: South China Agricultural University, 2017.(in chinese)
[34] 袁隆平. 超級雜交水稻育種研究新進展. 中國農村科技, 2010, Z1: 24-25.
YUAN L P. New progress in super hybrid breeding.China Rural Science & Technology, 2010, Z1: 24-25. (in Chinese)
[35] XIE Y Y, NIU B X, LONG Y M, LI G S, TANG J T, ZHANG Y L, REN D, LIU Y G, CHEN L T. Suppression or knockout ofovercomes the-mediated hybrid male sterility in rice. Journal of Integrative Plant Biology, 2017, 59(9): 669-679.
Genetic analysis and candidate gene identification on fertility and inheritance of hybrid sterility ofandcross
XU Na, TANG Ying, XU ZhengJin, SUN Jian, XU Quan
Rice Research Institute, Shenyang Agricultural University, Shenyang 110866
【Objective】The F1hybrid sterility between/and/severely hinders the utilization of hybrid advantage between subspecies. Exploring the genetic mechanism and identifying new regulatory genes for/hybrid sterility will provide theoretical basis for promoting genetic improvement of/hybrid seed setting rate.【Method】A series of stable genetic recombination inbred lines (RILs) containing 95 plant lines were derived from the cross betweenvariety Habataki andvariety Sasanishiki after 10 generations inbred using single seed descent method. High throughput sequencing was performed on both parents and RILs on the Illumina platform, and the distribution of Habataki pedigree in RILs was analyzed at the whole genome level. The segregation distortion regions were identified, and hybrid sterile related gene loci were screened within the segregation distortion regions, then identified candidate genes through sequence alignment comparison. The targeted gene was knockout to verify the function using CRISPR gene editing technology.【Result】The hybrid F1plants derived from the cross between Habataki and Sasanishiki showed significant heterosis in panicles, grains per panicle, and thousand grain weight, but its seed setting rate significantly decreased. I2-KI microscopy revealed a significant decrease in F1pollen fertility. High throughput sequencing of the entire genome of RILs revealed significant segregation distortion on chr.1, chr.3, chr.5, chr.6, chr.7, and chr.12, indicating that the genotype in this region tends towards the Habataki. Sequence alignment comparison revealed that,, andare target genes for the segregation distortion on chr.3, chr.6, and chr.12. The CRISPR gene editing mutants with a knock-outallele in Habataki successfully improved the pollen fertility and seed setting rate of F1hybrid with Sasanishiki. A complex structural variation was found between Sasanishiki and Habataki in the segregation distortion of chr.1. A 24.7 kb segment containing 4 predicted genes in the Sasanishiki genome was replaced by a 64.8 kb segment containing 10 predicted genes in Habataki, the structural variation may involve in controlling the hybrid sterility ofandcross. 【Conclusion】This study detected multiplehybrid infertility related loci, and successfully improved F1fertility by using CRISPR gene editing to knock out multiple copies ofin Habataki, locking in the target gene in theregion of chr.1.
rice; hybrid sterility; high throughput sequencing; gene editing;candidate genes

10.3864/j.issn.0578-1752.2024.08.001
2023-10-23;
2023-11-29
興遼英才計劃(XLYC2203171)
許娜,E-mail:xuna1109@163.com。通信作者徐銓,E-mail:kobexu34@syau.edu.cn。通信作者孫健,E-mail:sunjian811119@syau.edu.cn
(責任編輯 李莉)
