液相色譜--串聯(lián)質譜法測定茶葉中敵百蟲的不確定度評定
2024-03-08 14:49:20楊麗蓉段聯(lián)勃柯順川符海霞
廣東茶業(yè) 2024年1期
楊麗蓉 段聯(lián)勃,2 柯順川 符海霞
(1.武夷星茶業(yè)有限公司,福建武夷山,354300;2.福建省企業(yè)技術中心,福建武夷山,354300)
日常的測量過程中,受到人員重復性、器皿設備精密度、試劑濃度精確度、方法適宜性及環(huán)境條件波動等因素的影響,測量值往往在一個區(qū)間內波動。不確定度反映了測量值的分散程度。不確定度的評定能夠幫助人們了解這種波動的程度,掌握測量值的可信程度。通過對影響測量值的不同因素進行不確定度評定,發(fā)現(xiàn)主要的不確定度分量,從而尋找出測量方法的改進機會,縮小波動的范圍進而增加測量值的可信度。
敵百蟲是一種高效、速效、廣譜,低殘留的有機磷農藥,有熏蒸、胃毒和觸殺等功能,可高效防治稻米、小麥、蔬菜、茶樹、水果、桑樹以及棉花等經濟作物上的咀嚼式口器害蟲、家畜寄生蟲和衛(wèi)生害蟲等[1]。GB2763-2021中規(guī)定茶葉中敵百蟲最大殘留量為2mg/kg,但茶葉樣品基質復雜,含有生物堿、色素、茶多酚等一系列干擾成分,給敵百蟲的準確測定造成了較大的不確定性[2]。在現(xiàn)有的茶葉檢測中,不確定度評定的文獻中以農藥殘留[5-8]和重金屬[9-12]為主,但有關液相色譜-串聯(lián)質譜法(Liquid chromatography - mass spectrometry,LC-MS/MS)檢測茶葉中敵百蟲的不確定度評定研究較為少見。
本研究以JJF 1059.1-2012《測量不確定度評定與表示》和CNAS-GL006-2019《化學分析中不確定度的評估指南》為依據,分析和評定了GB 23200.13-2016測定茶葉中敵百蟲殘留量的不確定度,為評價茶葉中敵百蟲殘留量測定結果的準確性與可靠性提供了一定的技術依據。
1 材料與方法實驗部分
1.1 試驗材料
乙腈、甲苯:色譜純,西隴科學股份有限公司;甲醇:色譜純,天津市光復科技發(fā)展有限公司;CleanertTPT 固相萃取柱:10mL,2g,博納艾杰爾公司;微孔濾膜(尼龍):13mm×0.2um,江蘇綠盟科學儀器有限公司;敵百蟲標準物質:純度97. 2%,德國Dr.Ehrenstorfer 公司;實驗用水為優(yōu)普純水機制備的超純水:電阻率
1.2 試驗方法
1.2.1 標準溶液配制
標準儲備液(500mg/L):準確稱取折算純度后的敵百蟲標準物質25.72mg,加甲醇溶解定容至50mL。
標準中間液(50mg/L):用5mL 移液器移取2.5mL 標準儲備液至25mL 容量瓶中,加甲醇定容至刻度。
工作標準溶液(1.0mg/L):用1mL 移液器移取中間液0.5mL 至25mL 容量瓶中,加甲醇定容至刻度。
上機標液配制:用1mL 移液器分別移取100uL、200uL、400uL、800uL、1000uL 工作溶液,至5mL 容量瓶中,空白基質提取液定容至刻度,移取1mL 上機。
1.2.2 樣品測定
稱取1 0.0 g 試樣(精確至0.0 1 g)于5 0 m L 具塞離心管中,加入3 0 m L 乙腈溶液,以1 5 0 0 0 r/m i n 均質提取1 m i n,4 2 0 0 r/m i n 離心5 m i n,移取上清液至雞心瓶中。殘渣加3 0 m L乙腈,重復上述均質離心過程,將上清液合并至雞心瓶中;再向殘渣中加入2 0 m L 乙腈,重復均質離心過程,合并上清液至同一雞心瓶中,4 5 ℃旋轉蒸發(fā)至近干,氮吹至干。殘留物用5 m L 乙腈復溶待凈化。C l e a n e r t T P T 加入約2 c m 高無水硫酸鈉后,加入5 m L 乙腈-甲苯溶液,待液面到達無水硫酸鈉頂部時,迅速移取1 m L 復溶液至C l e a n e r t T P T 凈化柱上,并更換另一潔凈雞心瓶接收。用2 5 m L 乙腈-甲苯溶液洗脫,收集洗脫液于雞心瓶中,4 5 ℃水浴濃縮至約0.5 m L,轉至3 5 ℃下氮氣吹干,加入1 m L 乙腈-水溶液溶解殘渣,經微孔濾膜過濾后,供液相色譜-串聯(lián)質譜測定。進行平行試驗,采用標準曲線法定量,測定結果用平行測定的算術平均值表示。
1.3 測量不確定度的評定方法
1.3.1 數(shù)學模型
式中:
X—茶樣中敵百蟲的含量,單位:mg/kg;
C—標準工作曲線中得到的敵百蟲濃度,單位:mg/L;
V—樣液定容體積,單位:mL;
M—試樣質量,單位:g;
REP—總重復因子;
R—敵百蟲回收率;
f—樣品稀釋因子,f=5。
1.3.2 不確定度來源的確定和分析
不確定度來源:測量重復性引入的不確定度分量ur(REP);檢測樣品稱量過程引入的不確定度分量ur(M);儀器測定帶來的不確定度分量ur(LCMSMS);標準溶液配制及稀釋時引入的不確定度分量ur(C);標準曲線擬合引起的不確定度分量ur(L);樣液定容體積引起的不確定度分量ur(V);回收率帶來的不確定度分量ur(R);樣品的制備和分樣嚴格按照檢測標準要求執(zhí)行,因此樣品均勻性引入的不確定度可忽略不計[18]。由數(shù)學模型導出:
2 結果與分析
2.1 不確定度的評定
2.1.1 測量重復性帶來的不確定度分量ur(REP)
該分量含人員、環(huán)境、測量方法及儀器重復性等其它分量的貢獻。樣品加標進行10 次重復分析,加標濃度為0.03mg/kg。測量結果通過下式計算重復性引入的相對標準不確定度ur(REP),測量及計算結果見表1。

表1 測量重復性引入的相對標準不確定度u (REP)
式中:
u(REP)—重復分析的標準不確定度;
yi—第i 個平行樣的測量結果;
y —測量結果的平均值;
n—平行樣的個數(shù),n=10;
ur(REP)—重復性引入的相對標準不確定度。
樣品稱量時使用的電子天平精度為0.01g,每次樣品稱量均準確稱取10.00g,電子天平校準證書顯示稱量范圍的最大允許誤差為±0.05g,依據矩形分布(k=√3),則:
樣品稱量的標準不確定度u(M)=δ/√3=0.05/√3=0.0289(g),
試樣質量為10.00g,其示值引入的相對標準不確定度ur(M)=0.0289/10.00=0.0029。
2.1.3 液相色譜質譜聯(lián)用儀儀器測定帶來的不確定度分量ur(LCMSMS)
計量證書顯示定量重復性為1%,依據矩形分布,該設備定量重復性引入的相對標準不確定度:
2.1.4 標準溶液配制和稀釋過程引入的不確定度分量ur(C)
該不確定度分量包括標準物質純度引起的標準不確定度ur(p)、標準物質稱量引起的標準不確定度ur(m)、操作溫度引入的不確定度ur(T),實際操作與校正溫度相同,則溫度引入的不確定度可忽略、儲備液配制定容到50mL 引起的標準不確定度ur(V1)、配制中間標準液移取儲備液引起的標準不確定度ur(V2)、中間標準液定容引起的標準不確定度ur(V3)、配制工作溶液移取中間液引起的標準不確定度ur(V4)、工作溶液定容引起的標準不確定度ur(V5)、移取不同體積工作溶液配制工作曲線引起的標準不確定度ur(V6)、標準工作曲線定容引起的標準不確定度ur(V7),則有:
標準物質證書顯示,敵百蟲標準物質的擴展不確定度U=1.02%,純度p 為97.20%,包含因子k=2,則下式計算出標準物質純度帶入的相對標準不確定度ur(p)=0.00525。
稱量使用精度為0.01mg 的分析天平,每次稱量均準確稱取經純度折算后的質量,電子天平校準證書顯示,稱量范圍的最大允許誤差為±0.5mg,依據矩形分布(K=√3),則標準物質稱量帶入的相對標準不確定度ur(m)=0.5/(√3×25/p)=0.01123。
50mL 容量瓶校準證書顯示,容量瓶最大允誤差為±0.05mL,依據矩形分布(k=√3),則儲備液配制定容到50mL 帶入的相對標準不確定度ur(V1)=0.05/(√3×50)=0.00058。
配制中間標準溶液用5mL 移液器移取儲備液2.5mL,查5mL 移液器校準證書移取范圍的最大允許誤差為±0.5%,依據矩形分布(K=√3),則配制中間標準液移取儲備液引起的相對標準不確定度ur(V2)=0.5%×2.5/(√3×2.5)=0.00289。
中間標準溶液定容至25mL,根據25mL 容量瓶校準證書提供的信息,容量瓶最大允許誤差為±0.03mL,依據矩形分布(k=√3),則該定容過程引起的相對標準不確定度ur(V3)=0.03/(√3×25)=0.00070。
配制工作標準液移取中間液用1mL 移液器移取中間液0.5mL,查1mL 移液器校準證書移取范圍的最大允許誤差為±1.0%,依據矩形分布(k=√3),則該移取過程引起的相對標準不確定度ur(V4)=1.0%×0.5/(√3×0.5)=0.00578。
工作標準溶液定容至25mL,25mL 容量瓶校準證書顯示,25mL 容量瓶最大允差為±0.03mL,依據矩形分布(k=√3),則該定容過程引起的相對標準不確定度ur(V5)=0.03/(√3×25)=0.00070。
配制上機工作曲線標液移取過程使用1mL 的移液器移取100μL、200μL、400μL 、800μL、1000μL共5 次,查1mL 移液器校準證書在移取范圍小于500μLuL 的最大允許誤差為±2.0%,按矩形分布(k=√3),則單次不確定度為:
在移取范圍大于500μL的最大允許誤差為±1.0%,則單次不確定度為:ur(V800μL)=ur(V1000μL)=1.0%/√3=0.00578;
配制上機工作曲線標液移取過程引起的標準不確定度ur(V6)通過下式結算結果為0.02162。
配制上機工作曲線標液定容過程,各曲線點定容至5mL,共定容5 次,查5mL 容量瓶校準證書,5mL 容量瓶最大允許誤差為±0.02mL,依據矩形分布(k=√3),則單次不確定度為:u(V7)=0.02/(√3×5)=0.00231
則配制上機工作曲線標液定容過程引起的相對標準不確定度為:ur(V7)=√(5×u(V7)2=0.00517
則:標準溶液配制和稀釋時帶入的相對不確定度ur(C)通過下式計算,ur(C)=0.0263。
2.1.5 標準曲線擬合引入的不確定度分量ur(L)
采用混合標液稀釋,用空白基質配制成5 種相對含量的標準溶液(10μg/kg、20μg/kg、40μg/kg、80 μg/kg、100μg/kg),每種濃度測定1 次,上機分別測定,每個濃度點測1 次,獲得相應峰面積,以最小二乘法擬合,得到直線方程y=bx+a(a 為截距,b 為斜率)和相關系數(shù)r,并按下面公式計算出擬合線性的標準差SL和擬合曲線引入的相對不確定度ur(L),具體數(shù)據及計算結果見表2。擬合線性的標準差按下式計算:
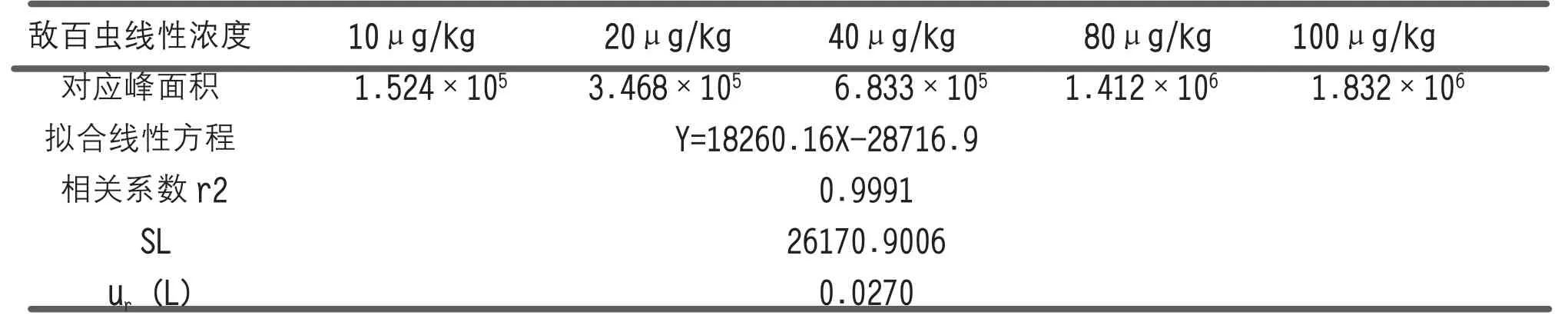
表2 標準曲線擬合引入的相對標準不確定度分量ur(L)
式中:
yij—農藥及相關化學品各濃度點的面積;;
m—每個濃度點的測量次數(shù);1 次
n—線性濃度點個數(shù);
SL—擬合曲線的標準差。
擬合曲線引入的不確定度u(L)按下式計算:
式中:
S_L—擬合曲線的標準差;
b—擬合曲線的斜率;
P—測試樣品的次數(shù);
n—測定標準溶液的次數(shù)(各濃度點之和);
x—未知樣的含量,單位μg/kg;
xi—標準曲線每個點對應含量,單位μg/kg。
則擬合曲線引入的相對不確定度ur(L)按下式計算:
2.1.6 樣液定容體積引起的不確定度分量ur(V)
用1mL(100-1000μL)移液器移取定容,其最大允許誤差為1.0%,依據矩形分布,k=√3,則:
吸取1mL 乙腈5 次溶解殘渣,則單次相對標準不確定度為:ur(V乙腈)=1.0%/√3=0.00578;
移取1mL 溶解物萃取時的相對標準不確定度為:ur(V溶解物)=1.0%/√3=0.00578;
吸取1mL 乙腈水時的相對標準不確定度為:ur(V乙腈水)=1.0%/√3=0.00578
則樣液定容過程引入的相對標準不確定度為:
2.1.7 回收率校正結果引起的不確定度分量ur(R)
表3 收集了匯總了近10 次日常添加的回收率及均值數(shù)據,1、2 組添加濃度為0.03mg/kg;3、4、5、6 組添加濃度為0.06mg/kg;7、8、9、10 組添加濃度為0.15mg/kg。回收率帶入的相對標準不確定度ur(R)通過下面兩式計算,計算結果見表3:

表3 回收率引入的相對標準不確定度u (R)
式中:
u(R)—回收率引起的標準不確定度;
Ri—第i 組測定的回收率結果;
R —回收率的平均值;
n—組別的個數(shù),n=10;
ur(R)—回收率帶入的相對標準不確定度。
并通過下式對平均回收率進行t 顯著性檢驗,以判定回收率在測量模式中是否需要采用以修正測定結果。在95%置信區(qū)間,自由度為9 時,t 分布臨界值為2.262,當t 檢驗值大于等于該臨界值時,說明存在顯著性差異,需要使用回收率進行結果校正,否則不需要。經計算檢驗值t=3.724,需要進行回收率校正。
2.2 合成標準不確定度的計算
按照以下公式可得合成標準不確定度為,經計算合成標準不確定度ur(X)=0.0522。
2.3 擴展不確定度U 的計算
取95%置信水平,k=2,敵百蟲含量測定的擴展不確定度根據以下公式計算,結果為U=0.0034。
2.4 不確定度報告
按照根據GB 23200.13-2016 規(guī)定的檢測程序進行,本次茶葉中敵百蟲含量測定的結果表示如下:敵百蟲含量=(0.0317±0.0034)mg/kg; k=2。
2.5 結語貢獻率
根據以下公式計算敵百蟲含量測定不確定度各分量的貢獻率根據以下公式計算,結果見表4。
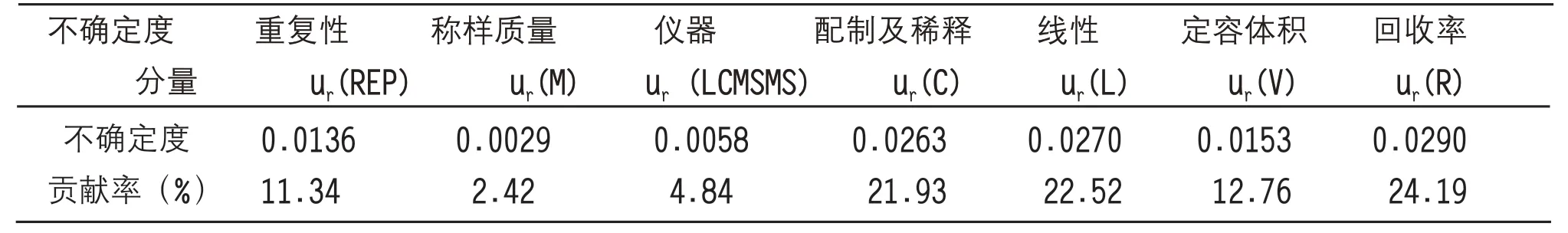
表4 不確定度分量的貢獻率
如表4 所示,通過比較不確定度各個分量的貢獻率顯示,敵百蟲檢測過程各環(huán)節(jié)對檢測結果的不確定度影響程度不同。過程中影響最大的是回收率、標準曲線最小二乘法擬合和標準溶液配制與稀釋過程的分量,其余因素按照從大到小依次為定容體積、測量重復性、液相色譜質譜聯(lián)用儀LC-MS/MS 儀器測定、樣品稱量質量。因此,按該標準進行敵百蟲檢測分析時,可通過將檢測結果進行回收率校正、加大標準曲線各濃度點的測定次數(shù)、使用精確度更高的移液管或其它移取設備進行標樣配制和定容以及增加檢測樣品平行測定的次數(shù)等手段來減少這些分量引入的不確定度,提高檢測結果的可靠性。
3 結論與討論
本文使用液相色譜-串聯(lián)質譜法對茶葉中敵百蟲的殘留量進行了分析,通過數(shù)學模型對不確定度的來源進行分析和確定后,分別對各不確定度分量進行了評定,最后通過不確定度分量貢獻率顯示:該方法中回收率帶入的不確定分量為最大不確定度。貢獻率的分析能夠為方法的優(yōu)化提供方向,從而通過合理優(yōu)化提高檢測的準確性。文章描述了不確定度評定的過程,為評價茶葉中農藥殘留測定的不確定度評定提供了一定的參考價值。

猜你喜歡
城市道橋與防洪(2022年4期)2022-07-01 06:04:12
中學生數(shù)理化·八年級物理人教版(2019年9期)2019-11-25 07:33:02
當代陜西(2019年8期)2019-05-09 02:22:48
中學生數(shù)理化·八年級物理人教版(2019年3期)2019-04-25 06:20:54
動漫星空(興趣百科)(2019年3期)2019-03-07 07:23:10
中學生數(shù)理化·八年級物理人教版(2018年3期)2018-05-31 08:52:45
家庭影院技術(2018年4期)2018-05-09 07:07:52
數(shù)學小靈通(1-2年級)(2017年10期)2017-11-08 08:39:45
少兒科學周刊·兒童版(2016年1期)2016-03-14 03:52:21
專用汽車(2016年4期)2016-03-01 04:13:43
