塞普替尼合成路線綜述
2024-03-05 14:07:20范昭澤
武漢工程大學(xué)學(xué)報 2024年1期
關(guān)鍵詞:方法
陳 龍,胡 偉,黃 璐,范昭澤*
1. 武漢九州鈺民醫(yī)藥科技有限公司,湖北 武漢 430071;
2. 武漢工程大學(xué)化工與制藥學(xué)院,湖北 武漢 430205;
3. 武漢大學(xué)藥學(xué)院,湖北 武漢 430072
抗癌藥物不斷被發(fā)現(xiàn),在腫瘤治療中發(fā)揮著越來越重要的作用[1]。腫瘤的靶向治療是一種針對特定靶向位點的細(xì)胞分子水平的治療方式[2]。2022 年7 月19 日,Eli Lilly 公司宣布,全球首個高選擇性的轉(zhuǎn)染重排(rearranged during transfection,RET)酪氨酸激酶抑制劑(RET 抑制劑)靶向藥物塞普替尼(selpercatinib,1)獲準(zhǔn)在中國海南博鰲超級醫(yī)院用于臨床急需,該藥物為RET 融合陽性的非小細(xì)胞肺癌(non-small cell lung cancer,NSCLC)和甲狀腺癌(thyroid cancer)患者在“先行先試”的政策條件下提供了新的治療選擇[3]。
塞普替尼研發(fā)代號為LOXO-292、LY352772,英文化學(xué)名為6-(2-hydroxy-2-methylpropoxy)-4{6-[6-[(6-methoxypyridin-3-yl)methyl]-3,6-diazabicyclo [3.1.1] heptane-3-yl] pyridin-3-yl}pyrazolo[1,5-α]pyridine-3-carbonitrile,中文化學(xué)名為6-(2-羥基-2-甲基丙氧基-4{6-[6-[(6-甲氧基吡啶-3-基)甲基]-3,6-二氮雜雙環(huán)[3.1.1]庚烷-3-基)吡啶-3-基]吡啶-3-基}吡唑[1,5-α]吡啶-3-甲腈,中文別名為賽哌替尼、塞普替尼、塞帕替尼、色普替尼、色普卡替尼等,其原研為美國Array 生物制藥(Array BioPharma)公司。2013 年7 月,Loxo Oncology Eli Lilly 公司與Array 公司簽訂多項許可和合作協(xié)議,由Loxo 公司進行后續(xù)研究開發(fā)。2020 年5 月8 日,美國FDA 批準(zhǔn)Eli Lilly 公司的口服塞普替尼膠囊(規(guī)格40 mg 和80 mg,商品名Retevmo)上市,用于治療:(1)轉(zhuǎn)移性RET 融合陽性的NSCLC 成人患者;(2)需要系統(tǒng)治療的晚期或轉(zhuǎn)移性RET 突變甲狀腺髓樣癌(medullary thyroid cancer,MTC)的成人和12 歲及以上兒童患者;(3)需要系統(tǒng)治療并且是放射性碘難治的晚期或轉(zhuǎn)移性RET 融合陽性甲狀腺癌的成人和12歲及以上兒童患者。塞普替尼是全球首個獲得批準(zhǔn)上市的專門針對攜帶RET 基因突變的癌癥患者的療法[4-10]。2022 年9 月30 日,Eli Lilly 公司的塞普替尼膠囊獲得NMPA 批準(zhǔn)在中國上市(規(guī)格為40 mg 和80 mg,商品名:睿妥)。
作者根據(jù)不同的起始物料和合成工藝綜述了塞普替尼的合成路線,匯總的合成路線圖見圖1。
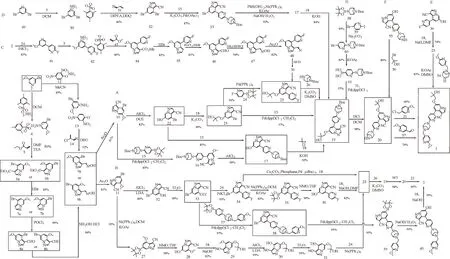
圖1 塞普替尼(1)的合成路線圖Fig.1 Graphical synthetic routes of selpercatinib(1)
1 方法A-C
此3 種方法均以3-溴-5-甲氧基吡啶(2)為起始物料。
1.1 方法A 和方法B
方法A 出自Array 公司提交的PCT 國際專利申請WO2018071454A1[11]中所述的合成路線和方法,MENDIOLA 等[12-17]分別對方法A 的部分或全部路線進行報道;方法B 出自Loxo 公司和Array公司提交的美國專利申請US20190106438A1[18]所述的合成路線和方法,此外David 等[19]對方法B的過程進行報道。
方法A 和方法B 分別以4-溴-6-甲氧基吡唑并[1,5-α]吡啶-3-甲腈(10)、6-溴-4-甲氧基吡唑并[1,5-α]吡啶-3-甲腈(11)作為關(guān)鍵中間體,依據(jù)多條合成路線制備目標(biāo)化合物塞普替尼(1)。化合物10 和化合物11 的合成過程相同,都是以3-溴-5-甲氧基吡啶(2)為起始物料,通過兩條路徑合成而來。化合物10 與化合物11 為區(qū)域異構(gòu)體,在合成此區(qū)域異構(gòu)體時,反應(yīng)條件及后續(xù)處理基本保持一致。
合成化合物10 和化合物11 的過程如下。
路徑一:以2 為起始物料,其與2,4,6-三甲基苯磺酰羥胺(3)發(fā)生氨基取代反應(yīng)得到1-氨基-3-溴-5-甲氧基吡啶-1-鎓-2,4,6-三甲基苯磺酸鹽(4),所得產(chǎn)物4 直接與丙炔酸乙酯(5)發(fā)生環(huán)合反應(yīng)分別得到4-溴-6-甲氧基吡唑并[1,5-α]吡啶-3-甲酸乙酯(6a)或6-溴-4-甲氧基吡唑并[1,5-α]吡啶-3-甲酸乙酯(6b),6a 或6b 在氫溴酸的作用下水解脫羧而得到4-溴-6-甲氧基吡唑并[1,5-α]吡啶(7a)或6-溴-4-甲氧基吡唑并[1,5-α]吡啶(7b),7a或7b 在三氯氧磷/DMF 條件下發(fā)生Vilsmeier-Haack甲酰化反應(yīng)得到4-溴-6-甲氧基吡唑并[1,5-α]吡啶-3-甲醛(8a)或6-溴-4-甲氧基吡唑并[1,5-α]吡啶-3-甲醛(8b),8a 或8b 與鹽酸羥胺發(fā)生肟化反應(yīng)得到4-溴-6-甲氧基吡唑并[1,5-α]吡啶-3-甲醛肟(9a)或6-溴-4-甲氧基吡唑并[1,5-α]吡啶-3-甲醛肟(9b),9a 或9b 經(jīng)與醋酐反應(yīng)后得到化合物10或化合物11。
路徑二:以2 為起始物料,與2,4-二硝基苯氧基胺(12)發(fā)生胺基取代反應(yīng)得到中間態(tài)產(chǎn)物1-氨基-3-溴-5-甲氧基吡啶-1-鎓-2,4-二硝基苯酚鹽(13),13 直接與2-氯丙烯腈(14)發(fā)生環(huán)合反應(yīng)得到化合物10 或化合物11。
1.1.1 方法A 化合物10 與3-(5-(4,4,5,5-四甲基-1,3,2-二氧戊環(huán)-2-基)吡啶-2-基)-3,6-二氮雜雙環(huán)[3.1.1]庚烷-6-羧酸叔丁酯(15)發(fā)生Suzuki 偶聯(lián)反應(yīng)生成3-(5-(3-氰基-6-甲氧基吡唑并[1,5-α]吡啶-4-基)吡啶-2-基)-3,6-二氮雜雙環(huán)[3.1.1]庚烷-6-羧酸叔丁酯(16),16 在無水AlCl3的作用下脫去甲氧基中甲基而生成3-(5-(3-氰基-6-羥基吡唑并[1,5-α]吡啶-4-基)吡啶-2-基)-3,6-二氮雜雙環(huán)[3.1.1]庚烷-6-羧酸叔丁酯(17),17 與1-氯-2-甲基-2-丙醇(18)在氫氧化鉀的作用下進行乙氧基化反應(yīng)得到關(guān)鍵中間體3-(5-(3-氰基-6-(2-羥基-2-甲基丙氧基)吡唑并[1,5-α]吡啶-4-基)吡啶-2-基)-3,6-二氮雜雙環(huán)[3.1.1]庚烷-6-羧酸叔丁酯(19),19在鹽酸的作用下脫Boc 得到4-(6-(3,6-二氮雜雙環(huán)[3.1.1]庚烷-3-基)吡啶-3-基)-6-(2-羥基-2-甲基丙氧基)吡唑并[1,5-α]吡啶- 3-腈(20),20 與5-(氯甲基)-2-甲氧基吡啶(21)發(fā)生N-烷基化反應(yīng)得到1。
化合物10 也可在無水AlCl3作用下脫去甲氧基中甲基成4-溴-6-羥基吡唑并[1,5-α]吡啶-3-甲腈(22)。通過22 有3 種路徑可以制備化合物19。
路徑一:22 與18 發(fā)生乙氧基化反應(yīng)生成4-溴-6-(2-羥基-2-甲基丙氧基)吡唑并[1,5-α]吡啶-3-甲腈(23),23 與2-氟-5-(4,4,5,5-四甲基-1,3,2-二氧硼雜環(huán)戊烷-2-基)吡啶(24)發(fā)生Suzuki偶聯(lián)反應(yīng)得到4-(6-氟吡啶-3-基)-6-(2-羥基-2-甲基丙氧基)吡唑并[1,5-α]吡啶-3-甲腈(25),25 與3,6-二氮雜雙環(huán)[3.1.1]庚烷-6-羧酸叔丁酯(26)發(fā)生N-烷基化反應(yīng)得到19。
路徑二:22 與18 發(fā)生乙氧基化反應(yīng)生成23后,23 可與15 發(fā)生Suzuki偶聯(lián)反應(yīng)得到19。
路徑三:22 與15 發(fā)生Suzuki 偶聯(lián)反應(yīng)得到17,17 與18 在氫氧化鉀的作用下進行乙氧基化反應(yīng)得到19,19 之后的反應(yīng)與上述制備1 的反應(yīng)過程一致。
1.1.2 方法B 化合物11 可通過4 種路徑制備目標(biāo)化合物1。其中有3 種合成路線需要合成至相同的關(guān)鍵中間體25,再按照方法A 中所述內(nèi)容來制備目標(biāo)化合物1。
合成化合物25 的方法如下。
路徑一:化合物11 在鈀催化劑作用下發(fā)生Miyaura 硼化反應(yīng)得到4-甲氧基-6-(4,4,5,5-四甲基-1,3,2-二氧硼雜環(huán)戊烷-2-基)吡唑并[1,5-α]吡啶-3-甲腈(27),27 在N-甲基嗎啉氮氧化物(NMO)作用下被氧化成6-羥基-4-甲氧基吡唑并[1,5-α]吡啶-3-甲腈(28),28 與18 發(fā)生乙氧基化反應(yīng)得到6-(2-羥基-2-甲基丙氧基)-4-甲氧基吡唑并[1,5-α]吡啶-3-甲腈(29),29 在無水AlCl3作用下脫去甲氧基中甲基成4-羥基-6-(2-羥基-2-甲基丙氧基)吡唑并[1,5-α]吡啶-3-甲腈(30),30 在三氟甲磺酸酯化試劑的作用下生成3-氰基-6-(2-羥基-2-甲基丙氧基)吡唑并[1,5-α]吡啶-4-基三氟甲磺酸酯(31),31 與24 發(fā)生Suzuki偶聯(lián)反應(yīng)得到25。
路徑二:化合物11 在無水AlCl3的作用下脫去甲氧基中甲基而生成6-溴-4-羥基吡唑并[1,5-α]吡啶-3-甲腈(32),32 在三氟甲磺酸酯化試劑的作用下生成6-溴-3-氰基吡唑并[1,5-α]吡啶-4-基三氟甲磺酸酯(33),33 與24 發(fā)生Suzuki 偶聯(lián)反應(yīng)得到6-溴-4-(6-氟吡啶-3-基)吡唑并[1,5-α]吡啶-3-甲腈(35),35 在鈀催化劑作用下發(fā)生Miyaura 硼化反應(yīng)而生成4-(6-氟吡啶-3-基)-6-(4,4,5,5-四甲基-1,3,2-二氧硼雜環(huán)戊烷-2-基)吡唑并[1,5-α]吡啶-3-甲腈(36),36 與18 在強堿的作用下發(fā)生乙氧基化反應(yīng)得到25。
路徑三:如同路徑二中方法,化合物11 合成至化合物34,34 在鈀催化及弱堿環(huán)境下與磷雜環(huán)丙烷類化合物及18 一鍋法合成25。
方法B 中第4 種合成化合物1 的過程是化合物11 按照上述路徑二中合成過程制備33 后,33 與6-[(6-甲氧基吡啶-3-基)甲基]-3-[5-(4,4,5,5-四甲基-1,3,2-二氧硼雜環(huán)戊烷-2-基)吡啶-2-基]-3,6-二氮雜雙環(huán)[3.1.1]庚烷(37)發(fā)生Suzuki 偶聯(lián)反應(yīng)得到6-溴-4-{6-[6-[(6-甲氧基吡啶-3-基)甲基]-3,6-二氮雜雙環(huán)[3.1.1]庚烷-3-基]吡啶-3-基}吡唑并[1 ,5-α]吡啶-3-甲腈(38),38 在鈀催化劑作用下發(fā)生Miyaura 硼化反應(yīng)得到4-{6-[6-[(6-甲氧基吡啶-3-基)甲基]-3,6-二氮雜雙環(huán)[3.1.1]庚烷-3-基]吡啶-3-基}-6-(4,4,5,5-四甲基-1,3,2-二氧硼雜環(huán)戊烷-2-基)吡唑[1,5-α]吡啶-3-甲腈(39),39在氫氧化鈉和過氧化氫的作用下被氧化成6-羥基-4-{6-[6-[(6-甲氧基吡啶-3-基)甲基]-3,6-二氮雜雙環(huán)[3.1.1]庚烷-3-基]吡啶-3-基}吡唑并[1,5-α]吡啶-3-腈(40),40 在強堿條件下與18 發(fā)生乙氧基化反應(yīng)得到1。
方法A 和方法B 都具有兩條合成路徑制備關(guān)鍵中間體10 或11,其中一條合成路徑需要進行6步反應(yīng)才能制備關(guān)鍵中間體,過于繁瑣,反應(yīng)得到6a 和6b[n(6a)∶n(6b)=1.6∶1],收率為46%~48%,其他反應(yīng)收率在80%~95%,總收率<20%,不適合工業(yè)化的需求;另一條合成路線只需3 步反應(yīng),大大優(yōu)化了反應(yīng)過程,提高了反應(yīng)收率。合成關(guān)鍵中間體10 或11 之后,方法A 和方法B 可通過多種路徑合成目標(biāo)化合物1,合成路線具有可選擇性,可以根據(jù)車間生產(chǎn)設(shè)備和生產(chǎn)成本進行路線選擇,更加適合車間規(guī)模生產(chǎn)需求,能較為快速地完成生產(chǎn)前準(zhǔn)備匹配市場需要。
方法A 的每一種路徑,都使用了昂貴的鈀試劑作為催化劑,可通過重復(fù)利用催化劑和溶劑套用的手段去降低反應(yīng)的物料成本,但是產(chǎn)品需要進行重金屬殘留的處理,增加了質(zhì)量研究難度,并且方法A 中有高溫、高壓的封管反應(yīng),危險性高,對反應(yīng)設(shè)備和操作人員的技術(shù)要求高。與方法A相比,方法B 具有反應(yīng)步驟少、反應(yīng)收率高等優(yōu)點。盡管方法B 中所有路線都使用鈀試劑,且少數(shù)步驟更是使用到過氧化氫,這使得反應(yīng)的成本以及反應(yīng)過程的危險性都有所增加,但是通過對鈀試劑和溶劑的重復(fù)套用可以大大減少成本并且站在工藝安全化的考慮,方法B 更容易工業(yè)化。
1.2 方法C
方法C 來源于江蘇慧聚藥業(yè)有限公司的中國專利申請CN202110688238.8[20]所公開的合成路線和方法。該方法是以2 為起始物料,經(jīng)多步反應(yīng)得到4-(6-氟吡啶-3-基)-6-甲氧基吡唑并[1,5-α]吡啶-3-甲腈(48),48 可用于制備塞普替尼(1)關(guān)鍵的中間體25。
該路線具體過程如下:2 與24 在鈀試劑作為催化劑的條件下發(fā)生Suzuki 偶聯(lián)反應(yīng)生成6'-氟-5-甲氧基-3,3'-聯(lián)吡啶(41),41 與3 反應(yīng)制備1-氨基-6'-氟-5-烷氧基-[3,3'-聯(lián)吡啶]-1-鎓-2,4,6-三甲基苯磺酸鹽(42),42 與丙炔酸甲酯(43)發(fā)生環(huán)合反應(yīng)得到4-(6-氟吡啶-3-基)-6-甲氧基吡唑并[1,5-α]吡啶-3-羧酸甲酯(44),44 在氫溴酸的作用下水解脫羧得到4-(6-氟吡啶-3-基)-6-甲氧基吡唑并[1,5-α]吡啶(45),45 在三氯氧磷/DMF 條件下發(fā)生Vilsmeier-Haack 甲酰化反應(yīng)得到4-(6-氟吡啶-3-基)-6-甲氧基吡唑并[1,5-α]吡啶-3-甲醛(46),46 與鹽酸羥胺發(fā)生肟化反應(yīng)得到4-(6-氟吡啶-3-基)-6-甲氧基吡唑并[1,5-α]吡啶-3-甲醛肟(47),47 與醋酐反應(yīng)得到48。用48 制備25 的過程為,48 在無水AlCl3作用下脫去甲氧基中甲基成36,36 與18 在堿性條件下發(fā)生乙氧基化反應(yīng)得到關(guān)鍵中間體25。
該方法主要提供了一種制備化合物48 的合成方法,48 是制備關(guān)鍵中間體25 的重要化合物。方法C 與方法A 及方法B 相同之處在于所用起始物料都為2;不同之處在于方法C 規(guī)避了原研發(fā)公司的化合物合成專利路線,創(chuàng)造性地設(shè)計一種合成25 的新工藝路線,但其過程中采用鈀試劑作為催化劑,使用了不易處理的鈀催化劑、極性與產(chǎn)物近似的丙炔酸甲酯和高腐蝕性的三氯氧磷和易揮發(fā)性的氫溴酸,且反應(yīng)后需要多次使用柱層析來純化產(chǎn)物,增加該方法的操作難度和生產(chǎn)成本,導(dǎo)致該方法難應(yīng)用于大規(guī)模生產(chǎn)中。
2 方法D
方法D 也來源于Loxo 公司和Array 公司提交的美國專利申請US20190106438A1[18]所述的合成路線和方法,David 等對方法F 的合成方法進行了后續(xù)報道[19]。
該方法是以3,5-二溴吡啶(49)為起始物料,經(jīng)7 步反應(yīng)制備化合物1。具體的過程如下:49 與3 反應(yīng)生成1-氨基-3,5-二溴吡啶-1-鎓-2,4,6-三甲基苯磺酸鹽(50),50 與丙烯腈(51)發(fā)生環(huán)合反應(yīng)生成4,6-二溴吡唑并[1,5-α]吡啶-3-甲腈(52),52與15 在鈀催化劑和弱堿的作用下發(fā)生Suzuki偶聯(lián)反應(yīng)生成3-[5-(6-溴-3- 氰基吡唑并[1,5-α]吡啶-4-基)吡啶-2-基]-3,6-二氮雜雙環(huán)[3.1.1]庚烷-6-羧酸叔丁酯(53),53 在鈀催化劑作用下發(fā)生Miyaura 硼化反應(yīng)及在氫氧化鈉和過氧化氫的作用下被氧化成17,17 的后續(xù)反應(yīng)按照方法A 中所述過程開展。
該方法是原研公司開發(fā)的另一種合成路線,其使用了不同的起始物料,進一步豐富了原研公司關(guān)于化合物1 的合成方法,與方法A 相比該方法具有路徑短、收率較高等優(yōu)點。與方法A 制備關(guān)鍵中間體17 過程相比較,方法D 同樣使用到昂貴的金屬催化劑和復(fù)雜的純化過程,但可通過重復(fù)套用及技術(shù)改進等優(yōu)化手段,可以降低方法D 的成本問題,并且方法D 的反應(yīng)條件相對溫和,使其更適合于工業(yè)化生產(chǎn)。
3 方法E
方法E 是武漢九州鈺民醫(yī)藥科技有限公司的中國專利CN201911346875.6[21]所述的合成路線和方法(該專利已獲得授權(quán))。該方法是以4-(6-氟吡啶-3-基)-6-羥基吡唑并[1,5-α]吡啶-3-甲腈(36)為起始物料,與18 在氫化鈉的作用下發(fā)生乙氧基化反應(yīng)生成25,25 與6-[(6-甲氧基吡啶-3-基)甲基]-3,6-二氮雜雙環(huán)[3.1.1]庚烷(54)發(fā)生N-烷基化反應(yīng)生成1。
該方法使用化合物36 作為起始物料,經(jīng)二步反應(yīng)制備化合物1,極大縮短了反應(yīng)的歷程,提高反應(yīng)的收率;該方法使用強堿NaH 替代原研工藝的弱堿作為乙氧基化反應(yīng)的催化劑,避免了高溫、高壓的危險反應(yīng)過程,但需注意大量使用氫化鈉時燃燒爆炸的危險。該方法具有路線短、總收率高和目前生產(chǎn)成本較高的特點,生產(chǎn)企業(yè)需要綜合性地考慮物料成本和時間成本之間的利弊。
4 方法F
方法F 是武漢九州鈺民醫(yī)藥科技有限公司的中國專利CN201911348328.1[22]所述的合成路線和方法(該專利已獲得授權(quán))。該方法是以4-[6-(3,6-二氮雜雙環(huán)[3.1.1]庚烷-3-基)吡啶-3-基]-6-羥基吡唑并[1,5-α]吡啶-3-腈(55)為起始物料,與1-溴-2-甲基-2-丙醇(56)在堿性試劑的作用下,進行醚化反應(yīng)生成20,20 與5-甲酰基-2-甲氧基吡啶(57)在氰基硼氫化鈉的作用下發(fā)生還原氨化反應(yīng)生成1。
該方法使用化合物55 作為起始物料,經(jīng)二步反應(yīng)制備化合物1,極大縮短了反應(yīng)的歷程,提高反應(yīng)的收率;該方法使用化合物56 替代原研路線中使用的化合物18,不僅規(guī)避了專利保護,也避免了高溫、高壓的危險反應(yīng)。該方法具有路線短、總收率高、避免高危險性化學(xué)反應(yīng)的優(yōu)點,但同方法E 一樣,該方法生產(chǎn)成本較高。
5 方法G
方法G 是察略盛醫(yī)藥科技(上海)有限公司提交的PCT 國際專利申請WO2020177668A1[23]中所述的合成路線。該方法是以3-(4,4,5,5-四甲基-1,3,2-二氧硼雜環(huán)戊烷-2-基)-3,6-二氮雜雙環(huán)[3.1.1]庚烷-6-羧酸叔丁酯(58)為起始物料,與5-溴-2-碘吡啶(64)在鈀試劑和碳酸鉀的作用下發(fā)生N-烷基化反應(yīng)生成3-(5-溴吡啶-2-基)-3,6-二氮雜雙環(huán)[3.1.1]庚烷-6-羧酸叔丁酯(59),59 在醋酸鉀及鈀試劑的作用下與聯(lián)硼酸頻那醇酯發(fā)生Miyaura 硼化反應(yīng)生成{6-[6-(叔丁氧基羰基)-3,6-二氮雜雙環(huán)[3.1.1]庚烷-3-基]吡啶-3-基}硼酸(60),60 在鈀試劑和碳酸鈉的作用下,與31 發(fā)生Suzuki 偶聯(lián)反應(yīng)生成19,后續(xù)反應(yīng)按照方法A 中所述過程開展。
該方法的優(yōu)點在于路線短和過程簡單,更加適合工業(yè)化生產(chǎn)的需求,缺點是多步反應(yīng)中使用鈀試劑作為催化劑,且多步反應(yīng)的后處理采用柱層析方式純化產(chǎn)物。綜上所述,該方法具有路線短、過程簡單、物料成本高和收率較低等特點,具有產(chǎn)業(yè)化的可能性。
6 結(jié) 論
本文綜述了采用5 種不同的起始物料、7 條不同的合成路線制備得到塞普替尼(1)的方法和過程。提出分別以3-溴-5-甲氧基吡啶(方法B)、3,5-二溴吡啶(方法D)和3-(4,4,5,5-四甲基-1,3,2-二氧硼雜環(huán)戊烷-2-基)-3,6-二氮雜雙環(huán)[3.1.1]庚烷-6-羧酸叔丁酯(方法G)為起始原料的3 條合成路線更具有工業(yè)化生產(chǎn)前景,與其他合成路線相比,該3 條合成路線的工藝原料易得,操作簡單,收率高,成本低。
猜你喜歡
中老年保健(2021年9期)2021-08-24 03:52:04
河北畫報(2021年2期)2021-05-25 02:07:46
中學(xué)生數(shù)理化(高中版.高考理化)(2020年2期)2020-04-21 05:33:04
兒童繪本(2020年5期)2020-04-07 17:46:30
兒童故事畫報(2019年5期)2019-05-26 14:26:14
Coco薇(2016年2期)2016-03-22 02:42:52
山東青年(2016年1期)2016-02-28 14:25:23
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年7期)2015-08-11 15:03:12
小雪花·成長指南(2015年4期)2015-05-19 14:47:56
