單層MoS2表面Fe,Ir摻雜對NO吸附與直接解離反應性能提升的理論計算
2024-01-15 00:21:58肖香珍胡林峰
吉林大學學報(理學版) 2024年1期
肖香珍, 胡林峰
(河南科技學院 化學與化工學院, 河南 新鄉 453003)
近年來, 研究人員采取多種方法研究二維材料在氣體傳感、 晶體管和催化劑等方面的性能[1-2]. 通過引入缺陷位和摻雜合適的金屬“雜質”能使二維材料的化學活性和傳感性能得到極大提高[3-6]. 二硫化鉬(MoS2)具有優良的導電導熱性能和較高的比表面積, 因而引起人們廣泛關注[7-13]. 單層MoS2具有對許多氣體敏感的特征, 目前已有較多的研究成果: Zhao等[14]利用密度泛函理論(DFT)發現NO和NO2能與具有較大吸附能的MoS2片發生強結合; 與單層MoS2相比, 摻雜的單層MoS2結構更能改善對氣體分子的吸附性能[15-17], 如Fe,Pt,Si,Ni等原子摻雜MoS2可以顯著改善MoS2的吸附性能[18-22]. 實驗結果表明, 利用低能氬濺射[23]或電子輻射可以在MoS2上控制產生S空位[10]. 理論上也探索了S空位具有作為催化活性位點的潛力[24]. 自然界中Fe的儲量豐富, 便宜易得, 且應用廣泛. Ir是一種稀有的貴金屬材料, 也是一種非常有潛力的多相反應催化劑, 在表面科學領域應用非常廣泛. 因此本文選擇非貴金屬Fe和貴金屬Ir這兩種過渡金屬摻雜MoS2的吸附和催化性能進行比較. 由于NO是一種有毒氣體, 帶有自由基, 化學性質非常活潑, 因此需要尋找氣敏性能較好的材料即時高效地檢測這種有毒氣體. 本文基于第一性原理方法, 對NO在兩種摻雜體系TM-MoS2(TM=Fe,Ir)表面的吸附和解離進行研究, 以期對進一步設計出廉價穩定的催化劑提供理論依據.
1 模型與計算方法
采用超胞(Slab)模型模擬MoS2表面摻雜Fe,Ir單原子的結構, Slab模型如圖1所示. 選取超胞大小為3×3, 由27個原子組成, 其中18個S原子, 8個Mo原子, 1個Fe原子或Ir原子. 圖1(B)~(E)分別是周期表面模型的俯視圖和側視圖. 由圖1可見, 俯視結構呈平面六角陣列方式排列, 具有二維層狀結構, 3個原子層中的原子呈夾心面包式排列, 即S—Mo—S, 摻雜原子放在表面一側.

圖1 Slab模型Fig.1 Slab model
文獻[25]計算了Fe,Ir吸附式摻雜和缺陷摻雜模型的吸附能和摻雜替換能, 發現S位摻雜體系更穩定, 實驗合成過程中也發現S空位缺陷更容易產生[10,14]. 摻雜后體系的穩定性由摻雜替換能ΔE分析:
ΔE=Ex+MoS2+EMo/S-Etotal-perfect-Ex,
(1)
其中ΔE為摻雜能, 其值越小體系越容易摻雜替換,Ex+MoS2為摻雜體系的總能,EMo/S為摻雜Mo或S原子,Etotal-perfect為單層超胞體系的總能量,Ex為摻雜原子的能量.
表1為Fe,Ir摻雜替換S,Mo原子的替換能. 由表1可見, 單層MoS2的S位置更容易被替換形成摻雜體系. 因此S位摻雜體系強于Mo位摻雜體系的穩定性. 因此計算均考慮將Fe,Ir單原子摻雜在S位置開展后續的研究工作.
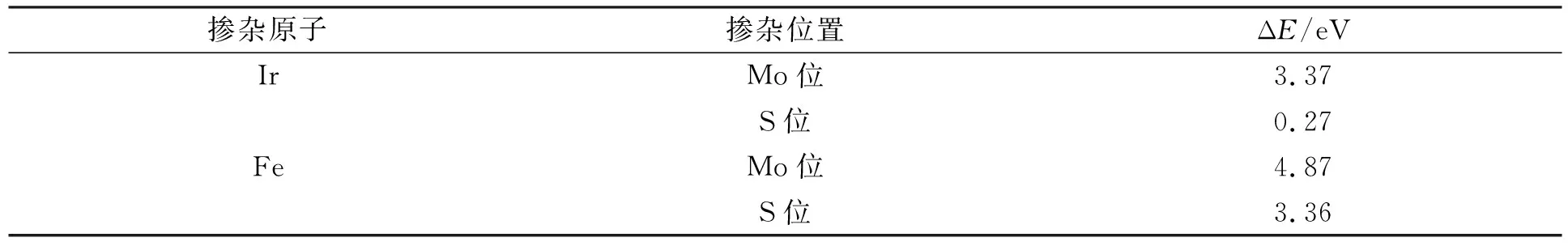
表1 Fe,Ir摻雜替換S,Mo原子的替換能
所有計算均基于贗勢平面波基組的密度泛函理論(DFT)[26-27], 采用vasp軟件包[28-29]完成. 選取廣義梯度近似下的PW91泛函(GGA-PW91)[30]描述體系的交換相關能, 采用PAW方法[31]描述離子芯勢, 設置平面波截斷能(ENCUT)為500 eV,K網格大小為5×5×1[32]. 平面上的所有原子均在三維方向上, 包括吸附的氣體小分子. 在優化過程中允許結構弛豫, 同時為充分模擬表面結構, 保證層間的作用不受影響, 平板間真空層厚度設為1.5 nm, 如圖1(A)所示. 當吸附構型的殘余應力小于1×10-4eV/nm時, 計算收斂[33]. 所有優化得到的最優構型均通過頻率分析加以驗證, 即局域極小點對應全部實頻, 無虛頻. 采用vasp軟件包的improved dimer method (IDM)[34-35]方法搜索反應過渡態、 確定最小能量路徑以及活化能, 對過渡態進行頻率驗證, 即過渡態有且只有一個虛頻[36], 且對應于相應鍵的伸縮振動模式.
2 結果與討論
為考察過渡金屬原子摻雜單層MoS2吸附性能和化學活性的影響, 將氣體分子NO以不同構型吸附在摻雜表面TM-MoS2(TM=Fe,Ir)的不同位置作為初始構型, 最終篩選得到最穩定的吸附構型. 氣相條件下, 將NO優化得到相應的鍵長分別為0.117,0.114 nm, 這些數據和文獻[37-39]結果一致. 氣體分子在摻雜表面的吸附能(Eads)表達式為
Eads=ENO/TM-MoS2-(ETM-MoS2+ENO),
(2)
其中ETM-MoS2,ENO和ENO/TM-MoS2分別表示摻雜體系的能量、 氣相分子NO的能量和氣體分子吸附在摻雜體系后的總能量. 吸附能越負, 吸附狀態越穩定.
2.1 NO在TM-MoS2(TM=Fe,Ir)體系表面的吸附
2.1.1 NO在TM-MoS2表面吸附的構型優化結果
圖2和圖3分別為Fe-MoS2和Ir-MoS2表面NO的吸附情況. 表2為NO分子在TM-MoS2(TM=Fe,Ir)表面吸附的幾何構型參數. 計算結果表明, 分子NO通過其N端與摻雜金屬原子(TM=Fe,Ir)分別以69.5°,87.9°形成了N—TM(TM=Fe,Ir)鍵, N—TM(TM=Fe,Ir)鍵長分別為0.165,0.179 nm, N—O鍵長分別為0.119,0.118 nm. 與氣態NO分子鍵長(0.117 nm)相比, 吸附在TM-MoS2(TM=Fe,Ir)表面的N—O鍵伸長約0.002,0.001 nm, 表明NO分子被輕微活化. 表3列出了NO和N2分別吸附在過渡金屬原子Fe,Ir摻雜單層MoS2表面的吸附能(Eads)、 解離活化能(Eact)以及反應熱(EΔH). 由表3可見, NO吸附在TM-MoS2(TM=Fe,Ir)表面相應的吸附能為-3.30,-3.17 eV, 吸附能較負, 表明Fe,Ir摻雜表面對NO有較好的吸附效果. 此外, 計算NO分子與完整MoS2表面間的吸附能為-0.05 eV, 吸附很弱, 導致后面的解離反應很難進行. 表明金屬原子摻雜缺陷位的改性方法可明顯提高單層MoS2表面對NO的吸附能力和催化能力, 摻雜Fe,Ir原子后MoS2對NO表現出較好的氣敏性能.

表2 NO分子在TM-MoS2(TM=Fe,Ir)表面吸附的幾何構型參數

表3 NO在TM-MoS2(TM=Fe,Ir)表面的吸附能、 解離活化能以及反應熱
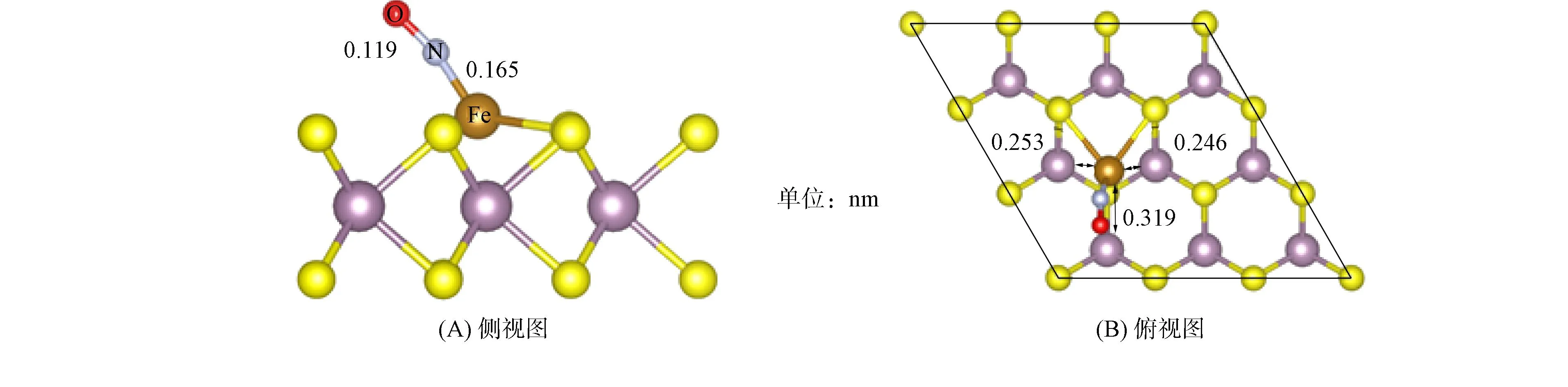
圖2 NO吸附在Fe-MoS2表面的側視圖(A)和俯視圖(B)Fig.2 Side view (A) and top view (B) of NO adsorbed on surface of Fe-MoS2

圖3 NO吸附在Ir-MoS2表面的側視圖(A)和俯視圖(B)Fig.3 Side view (A) and top view (B) of NO adsorbed on surface of Ir-MoS2
兩個摻雜體系中, 相鄰的Ir—Mo鍵的平均鍵長dIr—Mo為0.257 nm, 相鄰的Fe—Mo鍵的平均鍵長dFe—Mo為0.239 nm. 而吸附NO分子后, Fe與鄰近的3個Mo原子的距離分別為0.253,0.246,0.319 nm(圖2(B)), 與摻雜表面相比, Fe原子由于NO吸附而偏離原摻雜S的位置距離較大, 與鄰近的兩個S原子發生較強的相互作用, 形成了一種新的橋連結構, 使Fe與其中的一個鄰近Mo原子偏離距離達到0.079 nm, 進一步實現摻雜位點晶格的改變, 并顯示出了Fe的親硫性質. Ir與鄰近的3個Mo原子距離分別為0.290,0.256,0.293 nm(圖3(B)), 也有不同程度的改變, 表明NO與兩個摻雜表面存在一定的相互作用. 在改變摻雜位點附近結構方面, Fe摻雜優于Ir摻雜.
2.1.2 TM-MoS2表面吸附NO的電子結構
電荷密度差分圖可以反映吸附前后電荷密度的重新分布情況. 采用
ΔP=PNO/TM-MoS2-PTM-MoS2-PNO
(3)
計算電荷密度差, 其中ΔP表示體系的電荷密度差,PNO/TM-MoS2,PTM-MoS2和PNO分別表示吸附體系、 TM-MoS2摻雜催化劑表面以及吸附物NO的電荷密度.
圖4為NO吸附在TM-MoS2(TM=Fe,Ir)表面后體系的電荷密度差分圖, 其中紅色表示增加, 藍色表示減少. 由圖4可見, N原子與O原子中間以及摻雜Fe,Ir原子下方出現不同程度的電荷降低, 說明NO和摻雜原子Fe,Ir的部分軌道電荷發生了轉移. N原子與摻雜原子的中間出現電荷增加, 表明N原子與摻雜原子Fe,Ir之間存在強烈相互作用. 摻雜Fe,Ir原子附近主要表現為電荷減少, 說明吸附后, NO分子得到電子, 且所得電子主要來自表面的摻雜原子.

圖4 NO吸附在TM-MoS2(TM=Fe,Ir)表面后體系的電荷密度差分圖Fig.4 Charge density difference maps of system after NO adsorption on surface of TM-MoS2 (TM=Fe,Ir)
下面研究NO各軌道與體系TM-MoS2(TM=Fe,Ir)摻雜金屬原子Fe,Ir的各軌道相互作用關系, 計算并繪制兩個摻雜體系的分波態密度(PDOS)圖, 分別如圖5和圖6所示, 圖中Fermi能級均位于0處. 通過對體系態密度的分析可更具體了解吸附體系的電子結構以及吸附分子與表面的相互作用本質.

圖5 吸附前NO(氣態)(A), NO吸附在Fe-MoS2表面后(B)和NO吸附在Ir-MoS2表面后(C)投影至相應N原子的2s,2py,2pz和2px軌道的PDOSFig.5 PDOS of 2s,2py,2pz and 2px orbitals projected to corresponding N atoms before NO adsorption (gaseous state) (A),after NO adsorption on surface of Fe-MoS2 (B) and after NO adsorption on surface of Ir-MoS2 (C)

圖6 NO吸附在TM-MoS2(TM=Fe,Ir)表面后N與Fe原子(A)和Ir原子(B)的PDOSFig.6 PDOS of N and Fe atom (A), Ir atom (B) after NO adsorption on surfaces of TM-MoS2 (TM=Fe,Ir)
由圖5(A)和(B)可見, 與孤立的氣相NO分子態密度圖相比, 吸附后N的2py,2px和2pz均參與作用. 吸附后, N原子的各軌道DOS峰均不同程度地向Fermi能級方向移動. 特別是2py和2px軌道的峰由氣相狀態0處的尖峰雜化為0~2.5 eV處的平坦峰, 使NO能穩定地在該表面上吸附. 2pz軌道峰雜化較少, 約-7.5 eV處的尖峰吸附后能量幾乎不變, 但雜化為兩個鄰近、 強度略降低的尖峰和0~2.5 eV處平坦峰, 說明N原子的2pz軌道參與吸附作用較小. 類似的, NO在Ir摻雜MoS2表面的吸附是由于N原子與Ir原子成鍵, 吸附后N原子的2py,2pz,2px的態密度峰向高能級方向移動, 如圖5(C)所示. 2py和2px軌道在Fermi能級附近的尖峰變得平坦且寬, 能量約為0~2.5 eV, 說明這兩個軌道參與主要的成鍵作用. 2pz軌道也有較小的作用, 在約-7.5 eV處的尖峰稍有分裂.

2.2 TM-MoS2(TM=Fe,Ir)表面NO的解離
以NO吸附在摻雜Fe原子位置上最穩定結構(圖2)為初始反應物, 采用IDM方法搜索NO解離反應的過渡態, 所得過渡態構型(TS1和TS2)和相應解離生成物的結構(FS1和FS2)如圖7所示.
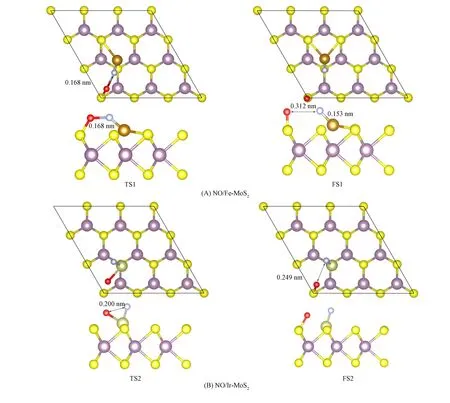
圖7 NO在摻雜TM-MoS2(TM=Fe,Ir)表面解離反應對應的過渡態和終態的結構示意圖Fig.7 Schematic diagram of transition and final states corresponding to dissociation reaction of NO on surface of doped TM-MoS2 (TM=Fe,Ir)
由圖7(A)可見, 在過渡態TS1中, N—O鍵的鍵長由初態的0.119 nm拉長為0.168 nm, 初態的N—O鍵以69.5°的角度傾斜結構擴散至N—O與表面幾乎平行的結構, Fe—N鍵的距離為0.158 nm, O原子與最近的S原子距離為0.169 nm. 該解離反應過渡態的虛頻為712.966 cm-1, 對應N—O鍵的伸縮振動. 在末態結構(FS1)中, N原子移動到與Fe原子稍近的位置, Fe—N鍵長為0.153 nm, Fe與鄰近的兩個S原子之間的距離分別為0.236,0.238 nm. O原子脫離N原子后吸附在最近的S原子上方, N與O原子之間的距離為0.312 nm. NO在Fe摻雜表面的解離活化能為3.22 eV, 整個解離過程吸熱1.77 eV, NO在Fe摻雜表面的吸附能為-3.30 eV, 因此在較高溫度下可以直接進行解離反應.
將NO吸附在摻雜Ir原子位置上最穩定結構為分解反應的初始結構(圖3), 計算所得過渡態結構, 由圖7(B)中TS2可見, N—O鍵長由初始結構的0.118 nm伸長至0.200 nm, 此時O原子向下傾斜, 與摻雜原子Ir距離拉近, N和O原子與Ir原子的距離分別為0.173,0.218 nm. 該反應的過渡態虛頻值為473.49 cm-1, 對應N—O鍵的伸縮振動, 該過程需要3.93 eV的解離活化能, 整個解離過程吸熱2.43 eV. 在末態結構(FS2)中, N—O鍵完全斷裂分離為1個N原子和1個O原子共吸附. N原子移動到與Ir原子附近形成Ir—N鍵, 鍵長為0.169 nm. O原子脫離N原子后吸附在最近的S原子上方, N與O原子之間的距離為0.148 nm.
相應的反應勢能曲線如圖8所示. 由圖8可見, 相對于貴金屬Ir, 較廉價的Fe摻雜后, NO分子在Fe摻雜體系高于Ir摻雜體系表面的吸附能, 且NO分解反應的活化能比在Ir-MoS2體系上低0.71 eV, 說明在Fe摻雜MoS2表面后, 其整體催化性能優于Ir原子摻雜.

圖8 NO在摻雜TM-MoS2(TM=Fe,Ir)表面解離反應的勢能曲線Fig.8 Potential energy curves of dissociation reaction of NO on surface of doped TM-MoS2 (TM=Fe,Ir)
3 結 論
綜上所述, 本文采用第一性原理方法分別對NO分子在TM-MoS2(TM=Fe,Ir)表面的吸附和解離行為進行了理論研究, 得到如下結論:
1) 與完整表面的物理吸附(-0.05 eV)不同, 在Fe,Ir摻雜MoS2表面NO吸附能分別為-3.30,-3.17 eV, 吸附能較大, NO在摻雜體系表面具有較好的吸附性能. 差分電荷密度計算結果表明, NO分子和摻雜原子Fe,Ir的部分軌道電荷發生了轉移, N原子與摻雜原子的中間電荷增加, 形成了共價鍵, 摻雜Fe,Ir原子附近電荷減少. 分波態密度分析表明, NO在摻雜MoS2表面的吸附是N原子與摻雜原子的部分軌道發生雜化.
2) 解離反應勢能曲線表明, 氣體NO在摻雜體系TM-MoS2(TM=Fe,Ir)表面的直接分解反應在摻雜表面均需較高能壘, 說明反應在較高溫度下才能發生, 可能在Ir摻雜的表面解離反應難以進行. 相對于貴金屬Ir, 摻雜較廉價的Fe后, 無論是吸附性能還是催化性能, Fe-MoS2摻雜體系表現出相對較好的性能, 為二維材料在催化性能以及氣體傳感性方面的應用提供了理論依據.
猜你喜歡
哲學評論(2021年2期)2021-08-22 01:53:34
新世紀智能(數學備考)(2020年11期)2021-01-04 00:38:16
中華詩詞(2019年7期)2019-11-25 01:43:04
中國外匯(2019年17期)2019-11-16 09:31:14
模具制造(2019年3期)2019-06-06 02:10:54
影視與戲劇評論(2016年0期)2016-11-23 05:26:01
現代企業(2015年9期)2015-02-28 18:56:50
現代企業(2015年1期)2015-02-28 18:43:18
新高考·高一物理(2014年1期)2014-09-18 01:26:07
土木建筑工程信息技術(2013年2期)2013-10-17 03:14:12
