第一代TRK抑制劑拉羅替尼關(guān)鍵手性胺中間體(R)-2-(2,5-二氟苯基)吡咯烷的合成工藝研究
2024-01-10 14:32:32裴超陳玉琴吳筱斐柴文靜楊愛青王磊磊劉團偉魏金建孫洪宜張志德
山東化工 2023年22期
裴超,陳玉琴,吳筱斐,柴文靜,楊愛青,王磊磊,劉團偉,魏金建,*,孫洪宜*,張志德*
(1.中瀚(齊河縣)生物醫(yī)藥科技有限公司,山東 德州 251114;2.山東師范大學化學化工與材料科學學院,山東 濟南 250014;3.齊魯工業(yè)大學(山東省科學院),山東省科學院生態(tài)研究所,山東省應(yīng)用微生物重點實驗室,山東 濟南 250014)
Tropomycin-related kinase (TRK)為神經(jīng)營養(yǎng)因子酪氨酸激酶受體,隸屬于受體酪氨酸激酶家族[1-3]。大量的研究表明TRK信號轉(zhuǎn)導通路的活化與腫瘤的發(fā)生、發(fā)展有很強的相關(guān)性,在神經(jīng)細胞瘤、前列腺癌、乳腺癌等中均發(fā)現(xiàn)了活化的TRK信號蛋白,近幾年來多種TRK融合蛋白的發(fā)現(xiàn)更顯示了其促進腫瘤發(fā)生的生物學功能[4-5]。如果能抑制激酶活性,就能抑制癌癥生長,TRK抑制劑的原理就是抑制激酶的活性[6]。
美國Loxo Oncology公司開發(fā)的第一代TRK抑制劑拉羅替尼(圖1a)可有效治療17種腫瘤,針對多種基因融合的患者有效率高達76%,包括12%的患者腫瘤完全消失,該藥于2018年被FDA批準上市,2022年4月13日獲得中國國家藥品監(jiān)督管理局批準正式上市[5-6]。拉羅替尼具有口服、針對17種不同腫瘤均可使用的廣譜靶向抗癌藥,臨床實驗結(jié)果證明拉羅替尼對不限年齡的TRK融合癌癥患者具有持久的抗腫瘤作用、良好的耐受性和很小的副作用等優(yōu)勢,是TRK基因突變癌癥患者的第一選擇[6]。然而拉羅替尼的價格非常昂貴,成人服用拉羅替尼膠囊30 d的費用約為32 800美元(約合人民幣20萬元),兒童服用其口服液的費用每月至少11 000美元。患者腫瘤部位的表面積越大,使用劑量越大,費用越高,一般患者根本望塵莫及,而科技的進步無法惠及萬千患者的根本在于原料藥的產(chǎn)業(yè)化工藝。拉羅替尼工業(yè)化的一個難點在于其關(guān)鍵手性胺片段(R)-2-(2,5-二氟苯基)吡咯烷(圖1b)的低成本制備,目前已報道的該中間體合成路線均通過手性誘導試劑或格氏試劑參與的反應(yīng)合成[7-10],通用的合成工藝路線如圖2所示。
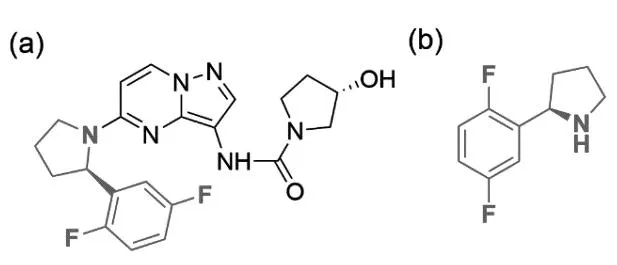
圖1 拉羅替尼和其關(guān)鍵手性片段(R)-2-(2,5-二氟苯基)吡咯烷的化學結(jié)構(gòu)式
路線1:
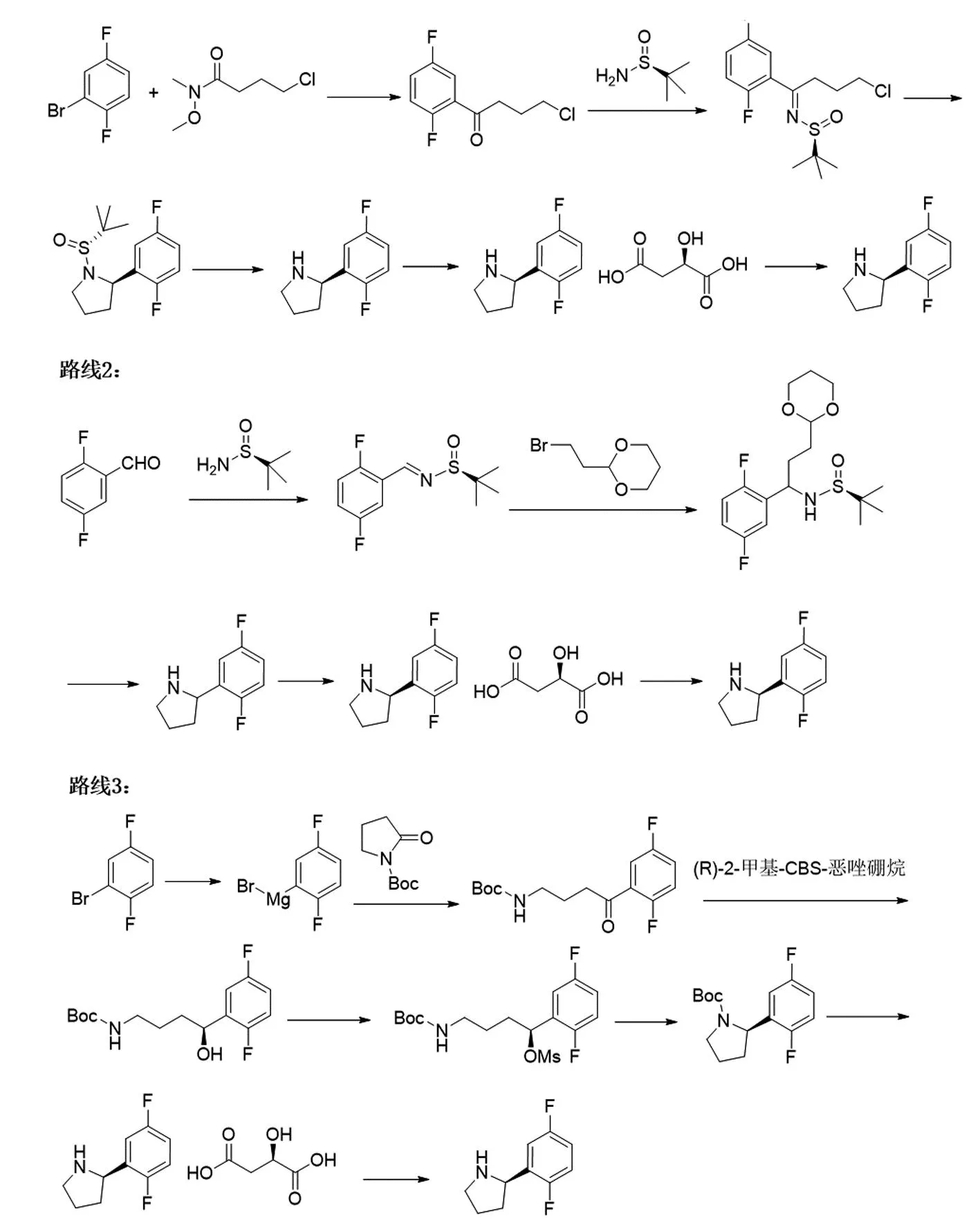
圖2 已報道的(R)-2-(2,5-二氟苯基)吡咯烷的合成路線
路線1和2均采用手性誘導試劑(S)-2-叔丁基磺酰胺合成手性胺中間體,該試劑昂貴且具有難聞的臭味,不符合現(xiàn)有的環(huán)保要求,難于規(guī)模化應(yīng)用;此外,路線1使用昂貴的三乙基硼氫化鋰(LiBEt3)還原脫除叔丁基亞磺酰亞胺,需在-78 ℃下進行,總收率僅為36%,生產(chǎn)成本極高,難以實現(xiàn)產(chǎn)業(yè)化[7];路線2使用昂貴的2,5-二氟苯甲醛及2-(2-溴乙基)-1,3-二氧雜環(huán)己烷,且需要危險的格氏反應(yīng)工藝,難于實現(xiàn)安全化,該路線原材料生產(chǎn)成本極高,難以規(guī)模化生產(chǎn)[8];路線3采用昂貴的手性試劑誘導手性中間體的合成,且用到硼烷二甲硫醚,具有難聞的臭味,不符合現(xiàn)有的環(huán)保要求,該路線也使用危險的格氏反應(yīng)工藝,難以實現(xiàn)操作安全化和規(guī)模化應(yīng)用[9-10]。以上報道的三條工藝路線雖然使用手性誘導催化劑,但所得的光學純度(ee值)較低,仍需對手性胺進行化學拆分才能達到≥98% ee值的質(zhì)量要求,生產(chǎn)成本極高。
為解決以上報道合成路線中存在的共性問題,本論文研究了拉羅替尼關(guān)鍵手性胺中間體(R)-2-(2,5-二氟苯基)吡咯烷的創(chuàng)新合成路線(圖3),該路線以廉價的2,5-二氟甲苯為起始原料,經(jīng)氧化得到中間體1,經(jīng)酯化生成中間體2,后經(jīng)親核取代、酸解、脫羧和環(huán)合一步法得到中間體3,后經(jīng)還原得到消旋化物中間體4,再經(jīng)化學拆分得到手性胺目標物,總收率為23%。該路線避免使用昂貴的原材料和試劑如手性誘導試劑、低溫和無水無氧環(huán)境,更適合拉羅替尼關(guān)鍵手性胺中間體(R)-2-(2,5-二氟苯基)吡咯烷的產(chǎn)業(yè)化。
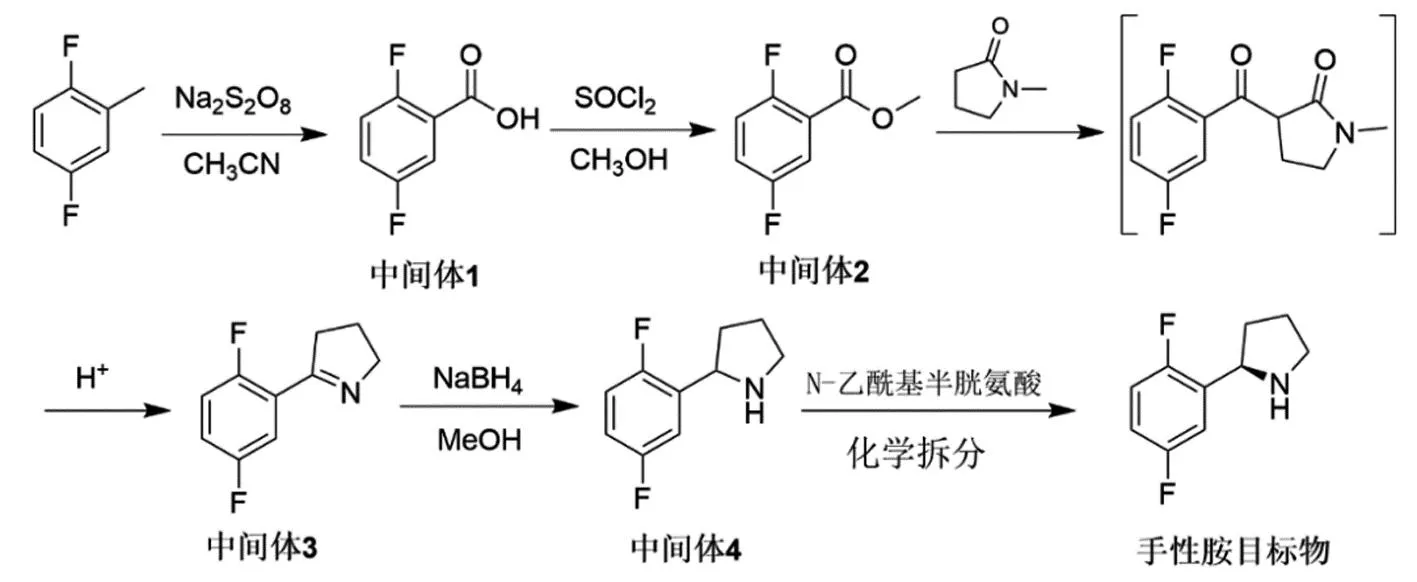
圖3 本論文報道的(R)-2-(2,5-二氟苯基)吡咯烷的新合成路線
1 實驗部分
1.1 儀器和試劑
400 MHz核磁共振譜儀(Bruker,Bruker Advance Ⅱ),高效液相色譜(Waters e2695)。
2,5-二氟甲苯、過硫酸鈉、乙腈、氯化亞砜、甲醇、N-甲基吡咯酮、鹽酸、硼氫化鈉、N-乙酰基-D-半胱氨酸均為分析純試劑,來源于國藥集團。
1.2 化合物的合成
1.2.1 2,5-二氟苯甲酸(中間體1)的合成
將2,5-二氟甲苯(20 g,0.16 mol)溶于100 mL乙腈中,室溫下緩慢加入過硫酸鈉(45.7 g,0.19 mol),緩慢升溫至60 ℃,TLC監(jiān)測至反應(yīng)完全(展開劑:正己烷),緩慢滴加水,析出淡黃色固體,抽濾、干燥得到23.5 g中間體1,收率95%,mp 132~134.1H NMR (400 MHz,d6-DMSO),δ: 13.54 (brs,1H,CO2H),7.60~7.63 (m,1H,Ar-H),7.51~7.55 (m,1H,Ar-H),7.37~7.42 (m,1H,Ar-H).19F NMR (372 MHz,d6-DMSO),δ: -116.07~-116.18 (m,1F),-118.04~-118.15 (m,1F)。
1.2.2 2,5-二氟苯甲酸甲酯(中間體2)的合成
將中間體1(22 g,0.14 mol)加入至110 mL甲醇中,室溫攪拌下緩慢滴加氯化亞砜(20.2 g,0.17 mol),滴加完畢后,升溫至70 ℃反應(yīng)5 h,TLC檢測反應(yīng)結(jié)束(展開劑:乙酸乙酯/甲醇,體積比100∶1),減壓蒸出溶劑,攪拌降溫至0~5 ℃,滴加飽和NaHCO3(aq)調(diào)節(jié)pH值=7~8,加入二氯甲烷萃取(100 mL×2),有機相干燥,減壓蒸干得到23 g中間體2,收率:95.8%。1H NMR (400 MHz,CDCl3),δ: 7.60~7.65 (m,1H,Ar-H),7.19~7.25 (m,1H,Ar-H),7.09~7.15 (m,1H,Ar-H),3.94 (m,3H,OCH3).19F NMR (372 MHz,d6-DMSO),δ: -115.42~-115.32 (m,1F),-118.12~-118.01 (m,1F)。
1.2.3 5-(2,5-二氟苯基)-3,4-二氫-2H-吡咯(中間體3)的合成
將N-甲基吡咯烷酮(13.8 g,0.14 mol)溶于100 mL乙腈中,降溫至-5~0 ℃,緩慢加入叔丁醇鉀(14.6 g,0.13 mol),攪拌10~15 min,將中間體2(20 g,0.12 mol)緩慢加入至上述反應(yīng)液中,滴加完畢后,室溫反應(yīng),TLC監(jiān)測中間體2消失(展開劑:乙酸乙酯/正己烷,體積比1∶5),降溫至室溫后,向上述反應(yīng)液中緩慢滴加15 mL濃鹽酸,升溫至70 ℃后保溫反應(yīng)6 h,調(diào)堿,減壓去除溶劑,二氯甲烷萃取、干燥,減壓除去溶劑得到淺黃色液體13.7 g,收率65.2%。1H NMR(400 MHz,CDCl3),δ: 7.22~7.27 (m,1H,Ar-H),6.91~6.97 (m,1H,Ar-H),6.83~6.87(m,1H,Ar-H),4.40(t,3H,J=7.6,CH2),3.13~3.22(m,1H),3.02~3.08 (m,1H),2.21~2.30 (m,1H),1.80~1.93 (m,1H),1.57~1.66 (m,1H).19F NMR (372 MHz,d6-DMSO),δ: -119.08~-118.98(m,1F),-124.87~-124.81 (m,1F)。
1.2.4 2-(2,5-二氟苯基)四氫吡咯(中間體4)的合成
將中間體3(13 g,0.07 mol)的甲醇溶液(130 mL)降溫至0~5 ℃,分批加入NaBH4(3.4 g,0.09 mol)后室溫反應(yīng),TLC監(jiān)控反應(yīng),反應(yīng)結(jié)束后,向體系中滴加水破壞NaBH4,減壓蒸出溶劑,二氯甲烷萃取,干燥有機相,蒸干得到12 g中間體4,收率92%。1H NMR(400 MHz,CDCl3),δ: 7.63~7.68(m,1H,Ar-H),7.02~7.10(m,2H,Ar-H),4.00~4.03(m,2H,CH2),2.70~3.02(m,2H,CH2),2.00~2.08(m,2H,CH2).19F NMR(372 MHz,d6-DMSO),δ: -118.89~-118.71 (m,2F)。
1.2.5 (R)-2-(2,5-二氟苯基)四氫吡咯(手性胺目標物)的合成
將中間體4(10 g,0.05 mol)加入到50 mL異丙醇中,加入N-乙酰基-D-半胱氨酸(8.1 g,0.05 mol),升溫80 ℃攪拌反應(yīng)5 h,降溫析晶,抽濾得到白色固體,將固體加入到水中,用2 mol/L NaOH(aq)調(diào)節(jié)堿性,二氯甲烷萃取,有機相干燥蒸干,重復(fù)上述操作一次,得到4.2 g淺黃色油狀手性胺目標物,收率42%,經(jīng)衍生方法HPLC檢測得ee值為99.5%。
1.3 (R)-2-(2,5-二氟苯基)吡咯烷的液相檢測方法
1.3.1 色譜條件
色譜柱:Xtimate?C18,4.6 mm×250 mm,5 μm P/N:00101-21043。
以0.01 mol/L乙酸銨/乙腈(體積比40∶60)為流動相,檢測波長為254 nm;流速為1.0 mL/min;柱溫為30 ℃;進樣量為10 μL。
1.3.2 溶液配制
1.3.2.1 制備溶液
向1.0 g(R)-2-(2,5-二氟苯基)吡咯烷中加入30%氫氧化鈉溶液,樣品溶液pH值≥14,加入甲叔醚劇烈搖勻,靜置分層,將上層溶液轉(zhuǎn)移至干凈的旋蒸瓶,加壓濃縮除去甲叔醚,作為制備溶液。
1.3.2.2 供試品溶液
取制備溶液10 mg,加入20 mg衍生試劑N-α-(2,4-硝基-5-氟苯基)-L-丙氨酸,加20 mL乙醇溶解,90 ℃下加熱回流2 min,濃縮除去乙醇,加入10 mL乙腈溶解,作為供試品溶液。
1.3.2.3 空白溶液
精密稱定20 mg衍生試劑N-α-(2,4-硝基-5-氟苯基)-L-丙氨酸,加20 mL乙醇溶解,90 ℃下加熱回流2 min,濃縮除去乙醇,加入10 mL乙腈溶解,作為空白溶液。
1.3.3 樣品測試
取供試品溶液、空白溶液,分別注入液相色譜儀,記錄色譜圖。按公司內(nèi)控標準,面積歸一化計,ee值應(yīng)不低于99.5%。
2 結(jié)果與討論
2.1 化合物的合成
與前期報道的(R)-2-(2,5-二氟苯基)吡咯烷工藝路線相比,本文報道的新路線避免了使用危險的格氏反應(yīng)、昂貴的手性誘導試劑、低溫和無水無氧操作。以廉價的起始原料2,5-二氟甲苯經(jīng)氧化得到中間體1,采用無機鹽過硫酸鈉作為氧化劑,避免使用了重金屬氧化劑,簡化了后處理步驟。第二步甲酯化采用傳統(tǒng)的SOCl2/MeOH方法,為除去少量的中間體1,反應(yīng)后處理需用弱堿如飽和NaHCO3溶液調(diào)劑pH值至7~8,pH值過高會導致中間體2的水解。本工藝路線的關(guān)鍵在于中間3的合成,借鑒最新報道的尼古丁合成路線,采用親核取代、酸解、脫羧和環(huán)合反應(yīng)一步法生成中間體3。N-甲基吡咯酮在強堿叔丁醇鉀的存在下,形成碳負離子,進攻中間體2羰基,形成1,3-二羰基化物,該中間態(tài)無需進行分離,加酸導致吡咯酮開環(huán),加熱條件下脫羧,而在酸性條件下,羰基和胺基發(fā)生分子內(nèi)環(huán)合生成中間體3。中間體4的合成采用經(jīng)典的NaBH4還原即可高效實現(xiàn),消旋體中間體4在N-乙酰基-D-半胱氨酸存在下經(jīng)化學拆分即可高效得到單一光學手性的(R)-2-(2,5-二氟苯基)吡咯烷。
2.2 核磁共振氫譜分析
中間體1的1H NMR顯示7.0~8.0為苯環(huán)上三個氫的特征峰,13.5為羧基氫特征峰,說明中間體1被成功合成;而中間體2的1H NMR 顯示除7.0~7.5為苯環(huán)上的三個氫的特征峰外,3.8為甲酯甲基氫特征峰,中間體11H NMR中對應(yīng)的13.5處羧基氫消失,說明甲酯化反應(yīng)成功;中間體3的1H NMR在2.0~4.0范圍內(nèi)為吡咯環(huán)的三組亞甲基氫特征峰,與中間體2的1H NMR進行對照,環(huán)合反應(yīng)發(fā)生且形成了中間體3;中間體4的1H NMR 在7.0~7.5為苯環(huán)上的三個氫的特征峰,4.2為與苯環(huán)相連的芐位氫特征峰,2.7為氨基氫特征峰,其他為吡咯環(huán)的三組亞甲基氫特征峰,說明還原反應(yīng)順利進行,且生成了中間體4,目標R型手性胺的1H NMR與中間體4相同。
2.3 純度分析
經(jīng)本文工藝路線合成的(R)-2-(2,5-二氟苯基)吡咯烷的HPLC譜圖如圖4所示,6.8 min處對應(yīng)目標R構(gòu)型,純度為99.82%,7.7 min為雜質(zhì)S異構(gòu)體,純度為0.18%,總體ee值為99.64%,遠大于現(xiàn)有技術(shù)中的98%ee值的質(zhì)量標準,完全符合現(xiàn)有的原料藥申報質(zhì)量規(guī)范對異構(gòu)體限度的控制要求。
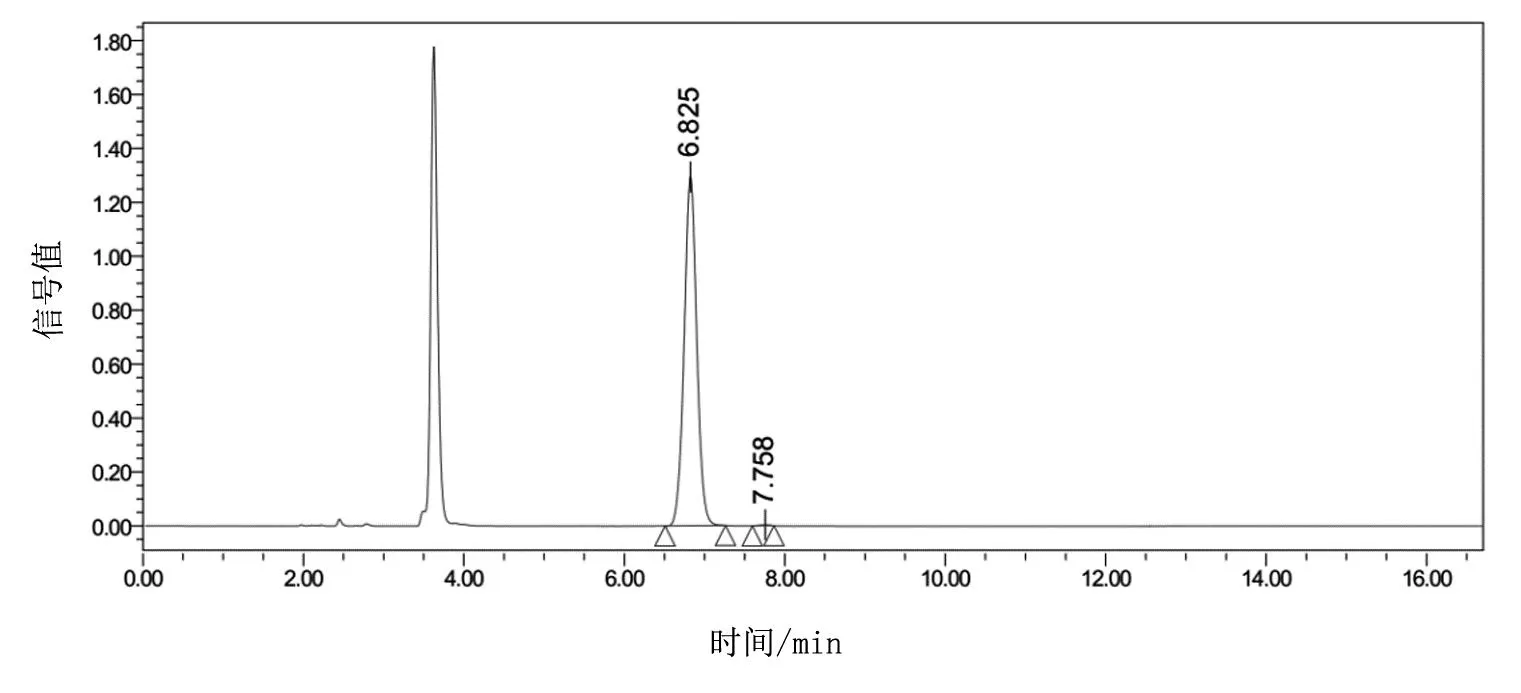
圖4 (R)-2-(2,5-二氟苯基)吡咯烷的HPLC檢測結(jié)果,6.825 min處對應(yīng)的
化合物為R型異構(gòu)體的純度,7.758min處對應(yīng)的雜質(zhì)為S型異構(gòu)體
3 結(jié)論
報道了拉羅替尼關(guān)鍵手性片段(R)-2-(2,5-二氟苯基)吡咯烷的新合成工藝路線,即以廉價的2,5-二氟甲苯為起始原料,經(jīng)氧化、甲酯化、后經(jīng)親核取代、酸解、脫羧和環(huán)合一步法得到亞胺環(huán)合物,經(jīng)還原得到消旋化物,再經(jīng)化學拆分得到手性胺目標物,總收率為23%,所得(R)-2-(2,5-二氟苯基)吡咯烷的光學純度為99.64%,遠大于現(xiàn)有技術(shù)中的98%ee值的質(zhì)量標準,完全符合現(xiàn)有的原料藥申報質(zhì)量規(guī)范對異構(gòu)體限度的控制要求。該路線避免了使用危險的格氏反應(yīng)、昂貴的手性誘導試劑、低溫和無水無氧操作,更適合拉羅替尼關(guān)鍵。
