茯神指紋圖譜結合化學模式識別和多指標成分定量研究
2024-01-09 00:46:12彭華勝張亞中劉軍玲金傳山
中草藥 2024年1期
關鍵詞:質量
王 娜,程 璐 ,王 浩 ,彭華勝, ,張亞中 ,劉軍玲 *,金傳山*
1.安徽中醫(yī)藥大學藥學院,安徽 合肥 230012
2.安徽省食品藥品檢驗研究院,安徽 合肥 230051
3.國家藥監(jiān)局中藥質量研究與評價重點實驗室,安徽 合肥 230051
4.中國中醫(yī)科學院 中藥資源中心 道地藥材品質保障與資源持續(xù)利用全國重點實驗室,北京 100700
茯神PoriacumPiniRadix為多孔菌科真菌茯苓Poriacocos(Schw.) Wolf 菌核中間天然抱有松根的白色部分,始載于《名醫(yī)別錄》[1],甘、淡,平,入心、脾經(jīng),具有寧心、安神、利水功效。茯神在臨床上仍是常用的大宗品種,《中國藥典》1963 年版[2]曾收錄“茯神”,然而,后來歷版藥典均未再收錄[3]。全國多個省已制定中藥材地方質量標準,但大多是基原、性狀、功效上的描述,內(nèi)容較少且不全面[4]。此外,通過查閱文獻資料,發(fā)現(xiàn)茯神相關文獻資料也較少。基于此,本研究采用高效液相色譜法[5-8]建立茯神的指紋圖譜[9-10],結合相似度評價同時進行化學模式識別[11-17],并測定其中10 個三萜成分的含量。從定性和定量2 個方面為該藥材質量標準的提升奠定基礎,保證茯神的臨床療效與用藥安全。
1 儀器與材料
Ultimate 3000 型高效液相色譜儀及所配備的二極管陣列檢測器(Dionex 公司);ML 型204 萬分之一電子天平(Mettler 公司);XP26 型百萬分之一電子分析天平(Mettler 公司);A11 型分析研磨機(IKA公司);KQ-100 型超聲儀(昆山市超聲儀器有限公司);XMTD205 水浴鍋(常州國宇儀器制造有限公司);Simplicity-185 型超純水儀(Millipore 公司);HALO 90 A C18色譜柱(250 mm×4.6 mm,5 μm)
對照品16α-羥基松苓新酸(批號J12HB184946,質量分數(shù)≥99%)、茯苓酸B(批號J13HB172712,質量分數(shù)≥95% )、去氫土莫酸( 批號A19GB158345,質量分數(shù)≥95%)、茯苓酸A(批號J28HB186446,質量分數(shù)≥96%)、多孔菌酸C(批號P08J11L107514,質量分數(shù)≥95%)、3-表去氫土莫酸(批號A26IB212331,質量分數(shù)≥95%)、3-O-乙酰基-16α-羥基松苓新酸(批號P08J11S107846,質量分數(shù)≥95%)、去氫茯苓酸(批號A19IB212921,質量分數(shù)≥98%)、松苓新酸(批號P24J10S80630,質量分數(shù)≥98%)、去氫齒孔酸(批號A18IB212947,質量分數(shù)≥95%)均購于上海源葉生物科技有限公司;乙腈、甲醇為色譜純,磷酸為分析純,水為超純水。
37 批茯神(S1~S17 批來自安徽,S18~S27批來自云南,S28~S37 批來自湖北),經(jīng)安徽省食品藥品檢驗研究院劉軍玲主任中藥師鑒定為多孔菌科真菌茯苓P.cocos(Schw.) Wolf 菌核中間抱有松根的白色部分。
2 方法與結果
2.1 色譜條件
采用HALO 90A,C18(250 mm×4.6 mm,5 μm)色譜柱,以乙腈(A)-0.05%磷酸(B)為流動相,梯度洗脫(0~9 min,62% A;9~28 min,62%~78% A;28~32 min,78%~90% A;32~38 min,90% A;38~40 min,90%~62% A),檢測波長242 nm,柱溫30 ℃,體積流量1.0 mL/min,進樣量10 μL。
2.2 溶液的制備
2.2.1 對照品溶液的制備 精密稱取16α-羥基松苓新酸、茯苓酸B、去氫土莫酸、茯苓酸A、多孔菌酸C、3-表去氫土莫酸、3-O-乙酰基-16α-羥基松苓新酸、去氫茯苓酸、松苓新酸、去氫齒孔酸對照品適量,加甲醇溶解,配制成質量濃度依次為0.038 3、0.049 0、0.048 5、0.033 7、0.047 0、0.043 0、0.038 4、0.054 7、0.089 6、0.040 9 mg/mL 的混合對照品溶液。
2.2.2 供試品溶液的制備 取本品粉末約1 g,精密稱定,置具塞錐形瓶中,精密加入甲醇25 mL,超聲處理(功率250 W、頻率40 kHz)30 min,濾過,濾液蒸干,殘渣用甲醇定容至5 mL 量瓶中,搖勻,濾過,即得。
2.2.3 陰性對照品溶液的制備 取甲醇,作為陰性對照品溶液。
2.3 HPLC 指紋圖譜方法學考察
2.3.1 專屬性試驗 取混合對照品溶液、供試品溶液(S2)和陰性對照溶液(甲醇溶液)分別按“2.1”項下色譜條件進樣測定。結果顯示,在色譜圖中各待測成分峰分離度良好,溶劑對色譜峰無影響,見圖1。
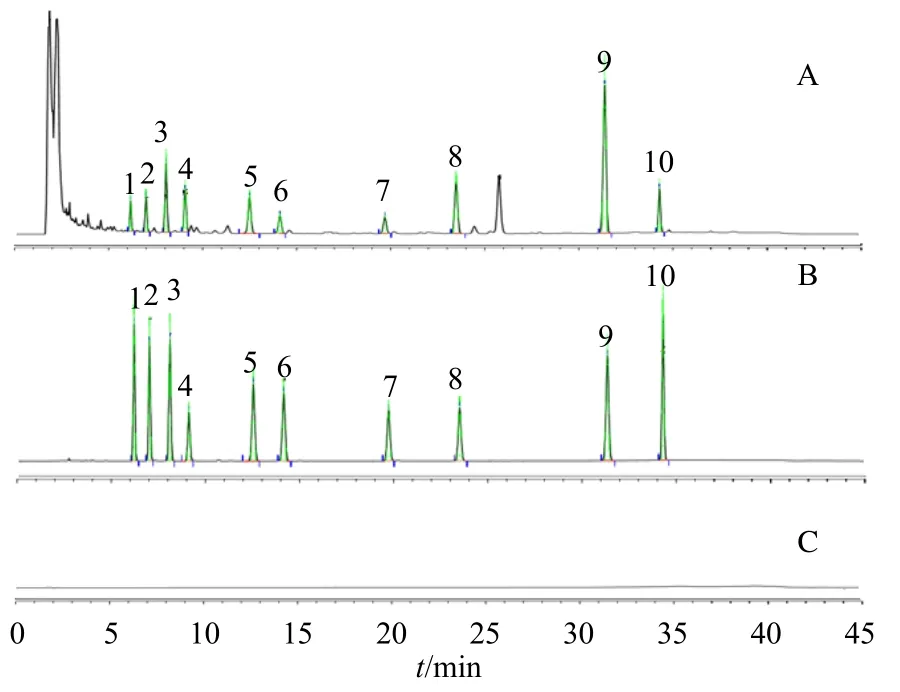
圖1 茯神供試品 (A)、混合對照品 (B) 和陰性對照品 (C)溶液的色譜圖Fig.1 Chromatograms of Poria cum Pini Radix test substance (A), mixed control substance (B), and negative control substance (C)
2.3.2 精密度試驗 取茯神樣品(S2),按照“2.2.2”項方法制備供試品溶液,按“2.1”項色譜條件重復進樣6 次,計算得到各共有峰相對保留時間及相對峰面積的RSD 均<1.5%,表明儀器精密度良好。
2.3.3 重復性試驗 取茯神樣品(S2),按照“2.2.2”項方法平行制備6 份供試品溶液,按照“2.1”項色譜條件進樣分析,計算得到各共有峰相對保留時間及相對峰面積的RSD 均<1.0%,表明該方法重復性良好。
2.3.4 穩(wěn)定性試驗 取茯神樣品(S2),按照“2.2.2”項方法制備供試品溶液,按“2.1”項色譜條件于制備后的0、2、4、6、12、24 h 進樣,計算各共有峰相對保留時間及相對峰面積RSD 均<2.0%,表明供試品溶液在24 h 內(nèi)穩(wěn)定。
2.4 指紋圖譜的建立及分析
2.4.1 指紋圖譜的建立 將所得的37 批茯神指紋圖譜依次導入到“中藥色譜指紋圖譜相似度評價軟件(2012 版)”。以樣品S1 作為參照圖譜,時間窗寬度設為0.1,采用中位數(shù)法,利用多點校正法進行峰匹配,生成對照圖譜,37 批茯神色譜圖與對照圖譜的疊加圖譜見圖2。
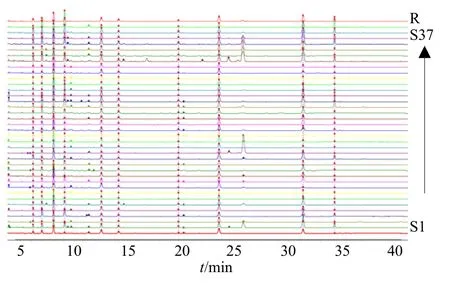
圖2 37 批茯神HPLC 疊加圖譜 (S1~S37)及對照圖譜(R)Fig.2 HPLC overlay spectra (S1?S37) and control spectra(R) of 37 batches of Poria cum Pini Radix
2.4.2 指紋圖譜的分析 37 批茯神指紋圖譜標定10 個共有峰,通過與對照品比對,指認出1 號峰為16α-羥基松苓新酸、2 號峰為茯苓酸B、3 號峰為去氫土莫酸、4 號峰為茯苓酸A、5 號峰為多孔菌酸C、6號峰為3-表去氫土莫酸、7號峰為3-O-乙酰基-16α-羥基松苓新酸、8 號峰為去氫茯苓酸、9 號峰為松苓新酸、10 號峰為去氫齒孔酸。計算37 批不同產(chǎn)地茯神之間的相似度,見表1,結果顯示,除S23、S37 外,其余35 批茯神樣品的相似度在0.791~1.000,S23、S37 可能由個體差異導致,其余樣品相似度較高。
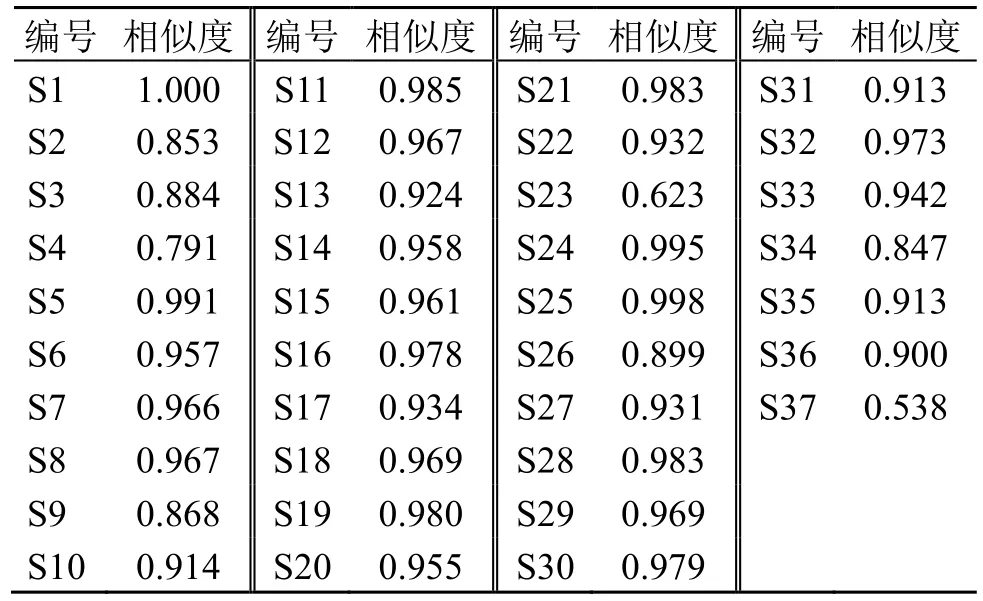
表1 37 批茯神相似度評價結果Table 1 Similarity evaluation results of 37 batches of Poria cum Pini Radix
2.5 多指標成分定量
2.5.1 線性關系考察 精密稱定16α-羥基松苓新酸、茯苓酸B、去氫土莫酸、茯苓酸A、多孔菌酸C、3-表去氫土莫酸、3-O-乙酰基-16α-羥基松苓新酸、去氫茯苓酸、松苓新酸、去氫齒孔酸的對照品適量,以甲醇配制成6 個不同質量濃度的系列混合對照品溶液,按“2.1”項下色譜條件進樣測定峰面積。以對照品濃度為橫坐標(X),對照品峰面積為縱坐標(Y)進行線性回歸分析,結果顯示各組分在相應濃度范圍內(nèi)線性關系良好。線性回歸方程、線性范圍及相關系數(shù)見表2。
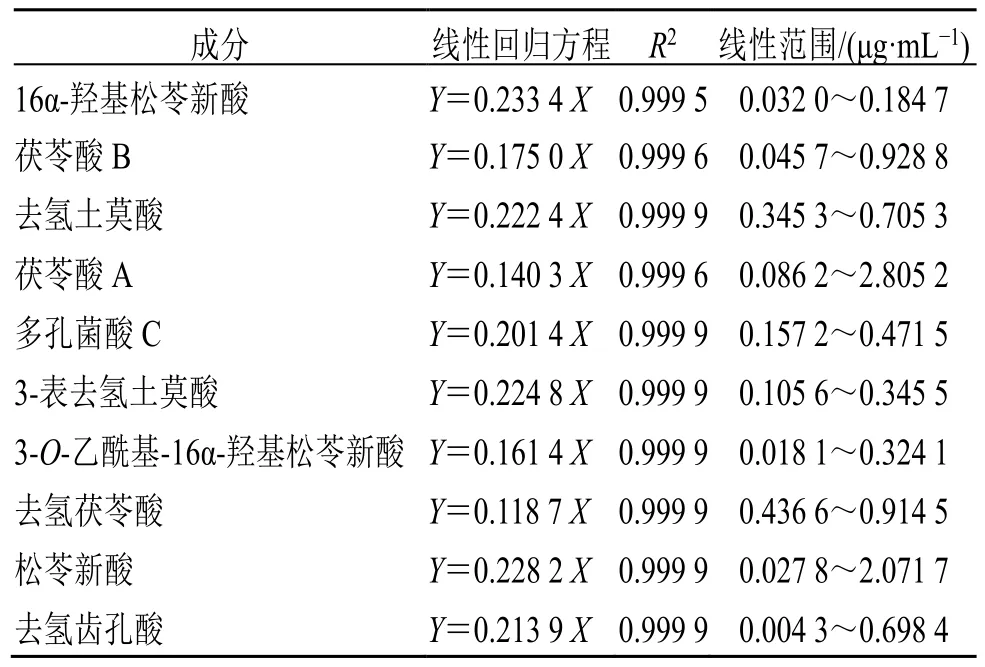
表2 茯神中10 種成分的線性回歸結果Table 2 Linear regression results of 10 components in Poria cum Pini Radix
2.5.2 精密度試驗 精密吸取“2.2.1”項下的混合對照品溶液,按“2.1”項下色譜條件連續(xù)進樣6 次,記錄峰面積,結果16α-羥基松苓新酸、茯苓酸B、去氫土莫酸、茯苓酸A、多孔菌酸C、3-表去氫土莫酸、3-O-乙酰基-16α-羥基松苓新酸、去氫茯苓酸、松苓新酸、去氫齒孔酸峰面積的RSD分別為0.001 6%、0.001 1%、0.001 9%、0.002 6%、0.003 9%、0.002 0%、0.001 7%、0.001 6%、0.002 4%、0.002 4%,表明儀器精密度良好。
2.5.3 重復性試驗 精密稱取茯神樣品粉末6 份(S2),按照“2.2.2”項方法制備供試品溶液,在“2.1”項色譜條件下平行進樣分析,結果16α-羥基松苓新酸、茯苓酸B、去氫土莫酸、茯苓酸A、多孔菌酸C、3-表去氫土莫酸、3-O-乙酰基-16α-羥基松苓新酸、去氫茯苓酸、松苓新酸、去氫齒孔酸質量分數(shù)的RSD 分別為0.002 8%、0.001 1%、0.001 4%、0.001 7%、0.013 4%、0.001 4%、0.001 5%、0.001 3%、0.001 1%、0.002 7%,符合實驗要求。
2.5.4 穩(wěn)定性試驗 精密吸取供試品溶液(S2),在“2.1”項色譜條件下,分別于0、2、4、6、12、24 h 進樣,計算出含量,結果16α-羥基松苓新酸、茯苓酸B、去氫土莫酸、茯苓酸A、多孔菌酸C、3-表去氫土莫酸、3-O-乙酰基-16α-羥基松苓新酸、去氫茯苓酸、松苓新酸、去氫齒孔酸質量分數(shù)的RSD 分別為0.002 3%、0.000 8%、0.000 6%、0.001 1%、0.017 2%、0.000 9%、0.001 5%、0.003 0%、0.000 7%、0.001 6%,樣品穩(wěn)定性良好,符合實驗要求。
2.5.5 加樣回收試驗 精密稱取已知成分含量的茯神樣品(S2)0.5 g 置錐形瓶中,平行6 份,按1∶1 比例加入各對照品,按“2.2”項下方法制備供試品溶液,在“2.1”項色譜條件下進樣分析,計算得到16α-羥基松苓新酸、茯苓酸B、去氫土莫酸、茯苓酸A、多孔菌酸C、3-表去氫土莫酸、3-O-乙酰基-16α-羥基松苓新酸、去氫茯苓酸、松苓新酸、去氫齒孔酸的平均加樣回收率分別為 90.27%、109.22%、97.90%、85.57%、107.41%、86.25%、88.45%、108.60%、96.22%、85.22%,RSD 分別為0.033 4%、0.016 1%、0.026 4%、0.024 6%、0.032 7%、0.029 7%、0.026 1%、0.030 3%、0.049 8%、0.036 8%,表明本方法的加樣回收率良好。
2.5.6 樣品含量測定 取37 批茯神藥材粉末各約1.0 g,精密稱定,按“2.2”項下方法制備供試品溶液,按“2.1”項下色譜條件測定,計算16α-羥基松苓新酸、茯苓酸B、去氫土莫酸、茯苓酸A、多孔菌酸C、3-表去氫土莫酸、3-O-乙酰基-16α-羥基松苓新酸、去氫茯苓酸、松苓新酸、去氫齒孔酸10個成分的含量。結果見表3。
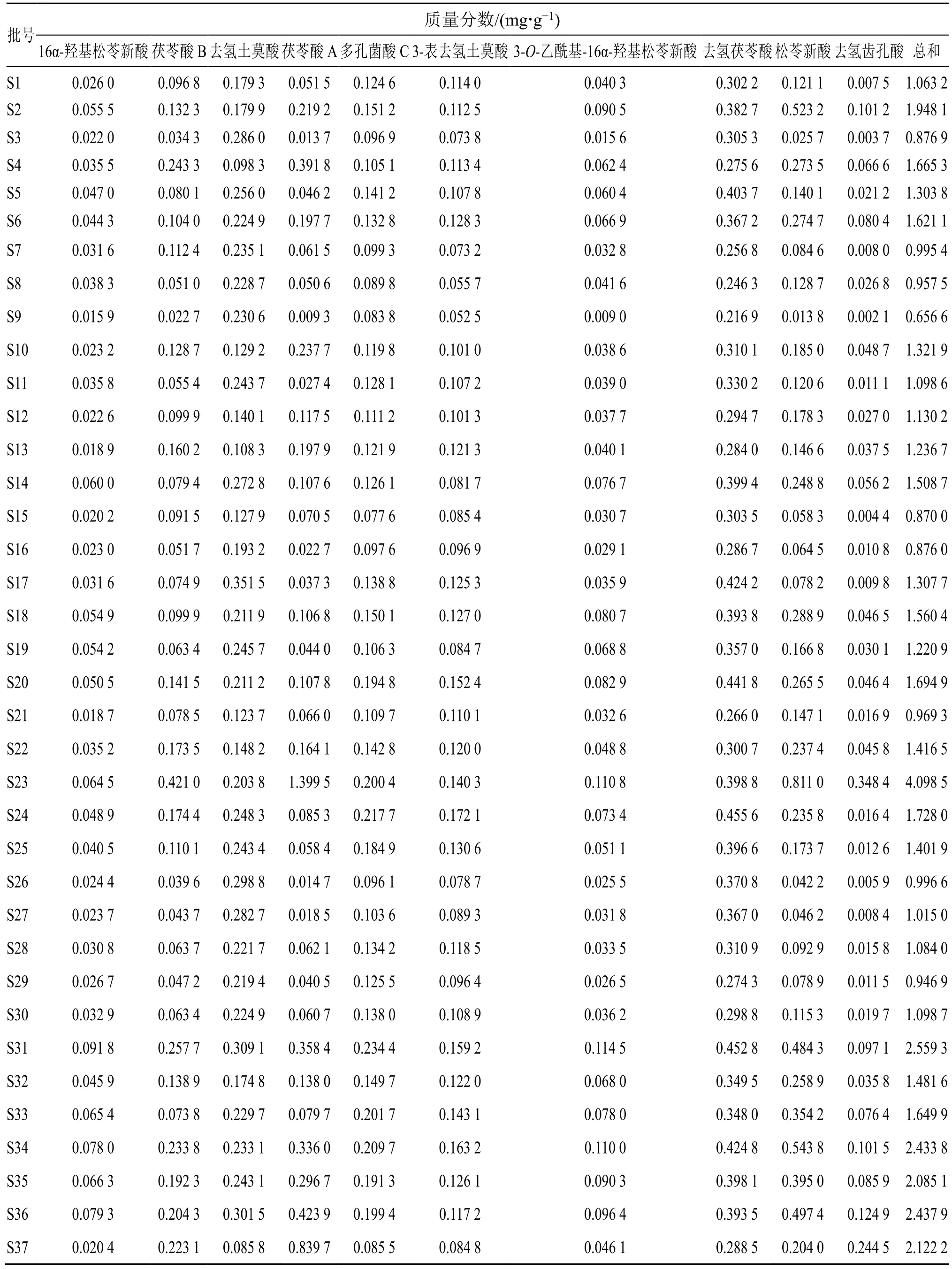
表3 樣品含量測定結果Table 3 Results of content determination of samples
2.6 化學模式識別分析
2.6.1 聚類分析 以37批茯神中10種化學成分的含量為變量,以平方歐氏距離為測距,運用IBM SPSS Statistics 23 軟件,采用系統(tǒng)聚類法,對37批茯神進行聚類分析,結果見圖3。當平方歐氏距離為5 時可分為3 類,S23 聚為一類,S37 聚為一類,其余樣品聚為一類,S23、S37 可能由個體含量差異導致,其余樣品間質量差異不大,與相似度結果相符。
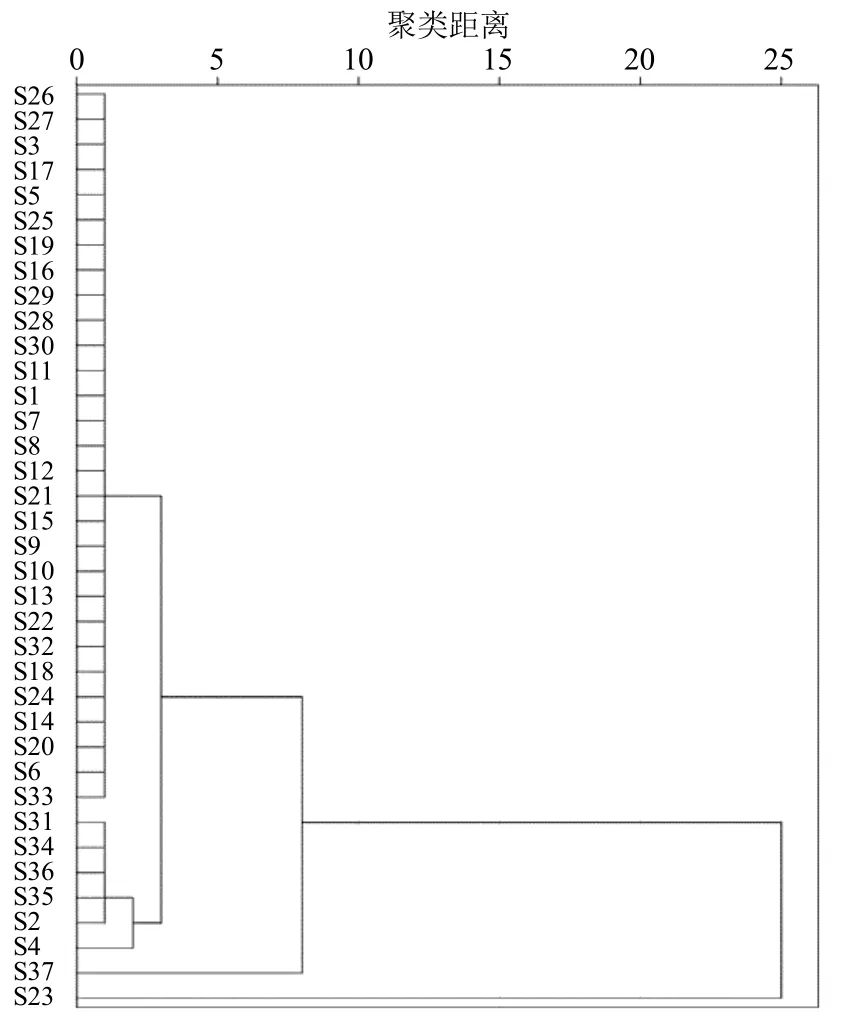
圖3 37 批茯神樣品的聚類分析圖Fig.3 Cluster analysis of 37 batches of Poria cum Pini Radix samples
2.6.2 主成分分析(principal component analysis,PCA) 為進一步比較不同批次茯神間的質量差異,將37 批茯神10 個有效成分含量導入IBM SPSS Statistics 23 和Simca 軟件,進行PCA,共有峰特征值見表4。前2 個主成分的初始特征值依次為6.098、2.248,累積方差貢獻率達到83.460%,表明以上2個主成分能夠較好的代表指紋圖譜中的大部分信息。PCA 得分圖顯示37 批樣品可大致劃分為3 類,與CA 結果一致,見圖4。

表4 主成分特征值及方差貢獻率Table 4 Principal component eigenvalues and variance contribution rate
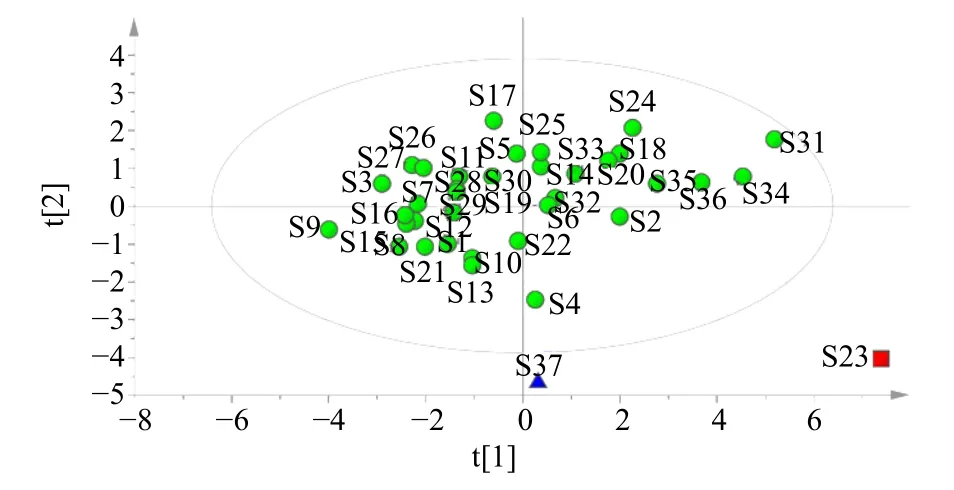
圖4 37 批茯神PCA 得分圖Fig.4 PCA score chart of 37 batches of Poria cum Pini Radix
2.6.3 正交偏最小二乘判別分析(orthogonal partial least squares discriminant analysis,OPLS-DA) 將37 批茯神有效成分含量數(shù)據(jù)導入到Simca 軟件進行OPLS-DA,以變量重要性投影(variable importance in projection,VIP)值>1.0 為標準進行篩選,結果見圖5,表明共有4 個特征峰的VIP 值>1.0,分別為峰8 去氫茯苓酸(VIP=1.775 9)、峰1 為16α-羥基松苓新酸(VIP=1.333 9)、峰5 為多孔菌酸C(VIP=1.125 1)、峰6 為3-表去氫土莫酸(VIP=1.107),表明這4 個成分是茯神不同批次樣品間質量差異貢獻較大的標志物,可以用來區(qū)分不同產(chǎn)地、批次間茯神的質量差異。OPLS-DA 分析的結果與CA、PCA 的結果一致。
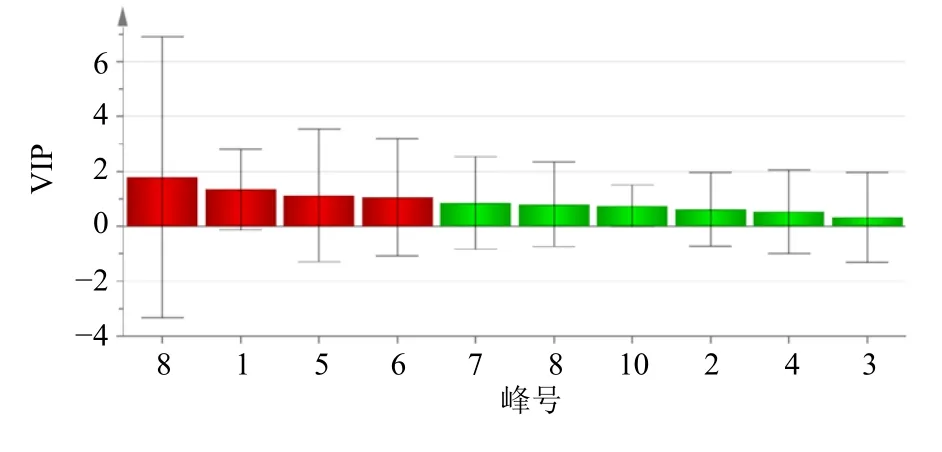
圖5 37 批茯神OPLS-DA 模型VIP 值圖Fig.5 VIP value chart of 37 Batch of Poria cum Pini Radix
3 討論
3.1 色譜條件及處理方法的選擇
本實驗在實驗操作過程中選用高效液相色譜法測定茯神三萜類成分,所用提取溶劑有乙醇、甲醇,提取方法有超聲與加熱回流,提取時間有20、30、40 min,發(fā)現(xiàn)甲醇超聲30 min 后提取的樣品溶液色譜峰信息較準確完整、操作簡單且高效節(jié)約,適用于茯神的定性及定量研究。在波長的選擇上,本文根據(jù)10 種待測成分的吸收波長特點,分別考察了在210、242 nm 波長下的檢測效果。結果發(fā)現(xiàn),在210 nm 波長下10 種三萜類成分均無吸收,在242 nm 波長下有最大吸收,故最終選擇242 nm 為測定波長。另外對流動相亦進行了考察,有乙腈-0.05%磷酸、乙腈-0.1%磷酸等。結果表明,以乙腈-0.05%磷酸進行梯度洗脫獲得的圖譜峰形良好,基線分布穩(wěn)定,可分別滿足對10 種三萜類化合物圖譜的分離要求。
3.2 指紋圖譜的建立
本實驗通過對37 批茯神藥材在相同的提取方法和色譜條件下所得HPLC 圖譜及數(shù)據(jù)進行分析,結合中藥色譜指紋圖譜相似度評價軟件(2012 版)分析結果,確定了茯神中共有15 個共有色譜峰,比對對照品色譜峰指認了16α-羥基松苓新酸、茯苓酸B、去氫土莫酸、茯苓酸A、多孔菌酸C、3-表去氫土莫酸、3-O-乙酰基-16α-羥基松苓新酸、去氫茯苓酸、松苓新酸、去氫齒孔酸,且在37 批樣品中均檢出這10 個成分。此外,部分樣品在26 min 左右檢出1個未知峰,這可能與茯神木的成分有關,有待進一步研究。除S23、S37 外,其余35 批茯神樣品的相似度在0.791~1.000,S23、S37 兩批樣品相似度存在較大差異,可能由個體差異導致,其余樣品相似度較高,表明茯神多批次產(chǎn)地的質量相對穩(wěn)定,能很好地反應樣品的指紋圖譜。
3.3 含量測定及化學計量學分析
本實驗通過CA、PCA 及OPLS-DA 分析對茯神含量進行綜合分析。通過CA 發(fā)現(xiàn),37 批茯神樣品可聚為3 類,S23 聚為一類,S37 聚為一類,其余樣品聚為一類,與相似度結果相符。對比表3 含量結果圖發(fā)現(xiàn),S23 中大多成分含量較高,而S37中大多成分的含量均較低,表明茯神中某些成分含量的高低可能是這兩批樣品各單獨聚為一類的原因,產(chǎn)地來源并不是決定聚類結果的唯一因素;PCA得分圖與CA 結果一致,利用主成分分析得到2 個主成分,可代表樣品中的大部分信息;此外,OPLS-DA 篩選出去氫茯苓酸、16α-羥基松苓新酸、多孔菌酸C、3-表去氫土莫酸這4 個質量差異標志物,表示這4 個色譜峰所代表的成分對區(qū)分不同產(chǎn)地、批次茯神的貢獻較大,可以用來區(qū)分不同批次間茯神的質量差異。
4 結語與展望
茯神是衛(wèi)生部頒布的第一批藥食同源兩用品[18],僅收載于地方標準,而且標準過于簡單,無法有效控制茯神質量,本研究建立的指紋圖譜及含量測定方法重復性良好,準確度高,可為茯神的質量控制提供新方法,彌補了現(xiàn)有質量標準的不足,可用于茯神的質量評價。此外,篩選出影響茯神質量的4 個潛在差異標志物,可為進一步完善該藥材的質量控制標準提供參考。
中藥材作為中醫(yī)用藥的源頭,其質量控制對后續(xù)中藥飲片和中成藥的安全性、有效性起到關鍵性作用,進而影響其臨床療效。通過市場調(diào)研,了解到目前市場流通的茯神有部分為后期在茯苓中人為插入松根而成,只為滿足茯神外觀性狀要求,而非真正的抱木而生,不具備茯神的功效;另外,以松根為中心,靠近松根多遠的范圍,算作真正意義上的茯神,暫無統(tǒng)一的標準,有待進一步研究,以確定其規(guī)格等級。因此,需進一步完善茯神質量標準體系,建立科學、管用的質量標準,服務于監(jiān)管,同時解決茯神質量摻偽問題。
利益沖突所有作者均聲明不存在利益沖突
猜你喜歡
中學生數(shù)理化·中考版(2022年10期)2022-11-10 09:37:42
中學生數(shù)理化·八年級物理人教版(2022年12期)2022-02-14 07:08:42
中學生數(shù)理化·八年級物理人教版(2021年12期)2021-12-31 03:23:08
中學生數(shù)理化·中考版(2020年10期)2020-11-27 01:59:48
中國生殖健康(2019年2期)2019-08-23 08:12:08
石油化工建設(2018年6期)2018-04-22 03:16:54
產(chǎn)品可靠性報告(2017年7期)2017-09-05 09:49:12
中學生數(shù)理化·八年級物理人教版(2017年12期)2017-04-18 12:59:38
汽車觀察(2016年3期)2016-02-28 13:16:26
民生周刊(2014年7期)2014-03-28 01:30:54
