代謝工程改造大腸桿菌發酵生產β-煙酰胺單核苷酸
2023-12-14 12:45:24安俊俠王倩倩王昭穎徐慶陽范曉光
食品科學 2023年22期
安俊俠,王倩倩,王昭穎,劉 歡,徐慶陽,范曉光
(天津科技大學生物工程學院,工業發酵微生物教育部重點實驗室,天津 300457)
β-煙酰胺單核苷酸(β-nicotinamide mononucleotide,β-NMN)屬維生素B族衍生物,廣泛存在于蔬菜、真菌、肉類和蝦類等天然食物中,參與人體內多種生化反應,與免疫、代謝調控息息相關[1-2]。在人體中,β-NMN作為輔酶I NAD+的前體,直接參與NAD+的合成,并通過NAD+體現其功能[3-4]。多項研究發現,β-NMN對緩解老年退行性疾病、神經退行性疾病、代謝紊亂和衰老等方面具有較好的作用[5-6]。β-NMN的食品安全性較高,日本、歐美等國家已經批準其作為新食品原料[7-8]。與NAD+相比,β-NMN更容易進入細胞,因此適合在食品、飲料、保健品中使用。
根據2013年我國第五次國家衛生服務調查分析報告中的老年人生活照料調查統計[1]數據顯示(以城市老年人生活照料分析為主),11.9%的在城市生活的老人在近30天的生活起居方面需要照顧時,49%由配偶照顧;47.4%由子女或孫子女照顧;0.8%由親戚、朋友、鄰居照顧;0.2%由社區提供照顧服務;1.8%是其他受助渠道獲取照顧服務或者是沒有人照顧,生活照顧需要自己獨立完成,具體見表1。
目前,β-NMN的生產方法主要分為化學合成法和酶催化法。化學合成法工藝成熟,是β-NMN的主要生產方法,但存在合成路線繁瑣、反應條件嚴苛、產物手性分離困難、使用有機溶劑等問題,導致產品售價較高[9-10]。酶催化法安全環保,產品旋光性單一,更容易被消費者接受[11]。酶催化法可以分為3 種路線,第1種是以煙酰胺核糖(nicotinamide riboside,NR)為底物,以ATP為磷酸供體,在煙酰胺核糖激酶的作用下生成β-NMN[12];第2種是以煙酰胺(nicotinamide,NAM)和5-磷酸核糖-1-焦磷酸(5-phosphoribosyl-1-pyrophosphate,PRPP)為底物,在煙酰胺磷酸核糖轉移酶(nicotinamide phosphoribosyl transferase,Nampt)的作用下生成β-NMN[13];第3種是以煙酸和PRPP為底物,在煙酸磷酸核糖轉移酶的作用下先生成煙酸單核苷酸(nicotinic acid mononucleotide,NaMN),然后在β-NMN合成酶的作用下生成β-NMN[14-15]。酶催化法的問題在于原料成本較高,酶活性和轉化效率較低,因此限制了工業級應用放大[16]。
大部分微生物可以通過自身代謝合成PRPP,因此理論上通過系統代謝改造,可以實現以NAM或煙酸為前體發酵生產β-NMN[17]。本研究以生長周期較短、遺傳背景清晰的野生型大腸桿菌(Escherichia coli)W3110作為出發菌株,設計模塊化代謝改造策略(圖1)。首先,分析并減弱底盤細胞中與NAM和β-NMN降解相關酶的表達,減少底盤細胞對前體和產物的額外消耗;其次,引入NAM輸入蛋白(BcNiaP)、β-NMN輸出蛋白(BmPnuC)、PRPP合成酶(PRPP synthetase,Prs)和Nampt,敲除調節蛋白PurR,實現了β-NMN的初步積累;此后,考察了不同來源的Nampt,篩選得到了酶活性較高且對底盤細胞負擔較小的催化用酶;最后,強化β-NMN輸出蛋白和Prs的表達水平,大幅提升了β-NMN的發酵產率。本研究通過基因組編輯和組成型質粒表達方式,構建1 株遺傳背景清晰、無營養缺陷、無需誘導的大腸桿菌基因工程菌,以期實現NAM為前體發酵生產β-NMN。

圖1 代謝工程改造大腸桿菌發酵生產β-NMN策略Fig.1 Metabolic engineering strategies for β-NMN fermentation in E.coli
1 材料與方法
1.1 材料與試劑
1.1.1 菌株與質粒
綜上所述,經過長期的發展,政府引導基金在撬動社會資本、政府意愿體現、政府參與公司治理能力和多階段投資引導能力等方面為新興產業發展進行了以FOF為代表的制度創新,但現有模式在考慮新興產業融資的多階段性、行業不對稱性等方面仍有較大不足,推動新興產業發展需要對融資特征進行準確定義,并在此基礎上進一步構建創新融資模式。
本研究使用的菌株和質粒分別見表1、2。其中,E.coliDH5α用于質粒構建,E.coliW3110 ΔlacI作為出發菌株用于構建β-NMN生產菌株。
翻轉課堂是常用的教學方法,高中物理實驗教學過程中,也采用了翻轉課堂的方法,這種教學方法相比于傳統的教學方法有很多優點.首先,它把課堂的大多數時間都歸還給學生,學生成為課堂的重心.翻轉課堂的提出,改變了傳統的授課模式,最大限度劃分課堂時間,合理配置教學資源,更好地為學生服務.這種教學模式在高中物理實驗中的應用,不僅提高了學生的學習效率,而且還促進了教育事業的改革.

表1 本研究所用菌株Table 1 Strains used in this study

表2 本研究所用質粒Table 2 Plasmids used in this study
1.1.3 引物
業務流程優化指中小出版企業通過自主研發和外購系統,實現采、編、印、版、售等技術創新,聚焦于出版產業全鏈數字化輔助技術,促進綠色印刷業務的開拓。“四維傳媒”基于“云計算”,將數字出版技術與移動平臺技術順利嫁接,形成每個在線服務技術模塊,實現遠程創意、中央圖庫、在線編輯、遠程批注、色彩管理、遠程打樣、綠色印刷以及在線全媒體閱讀等全鏈整合,極大地提高了數字出版效率。截至2017年年底,“四維傳媒”的“綠色出版”系統已經獲得多項海外機構認證,并與歐美4個國家建立了戰略合作伙伴關系。“龍源數媒”不斷創新和迭代觸控閱讀和互聯網技術,提高了產品“按需出版”的能力,為新增公共文化產品線預留了出版空間。
在細胞中實現β-NMN的發酵生產,需要考慮3 個方面的問題,一是前體NAM和產物β-NMN的轉運;二是前體PRPP的供應;三是催化β-NMN合成關鍵酶Nampt的活力。Shoji等[27]發現Burkholderia cenocepacia來源的煙酸轉運蛋白(BcNiaP)以及Bacillus mycoides來源的NR轉運蛋白(BmPnuC)可以分別提高大腸桿菌對NAM的攝取和β-NMN的外排。BcNiaP和BmPnuC均為膜蛋白,表達過強將會影響菌體生長,因此將niapBc和pnuCBm基因整合至工程菌N-8基因組ilvG和ygaY位點上,并使用組成型Ptrc啟動子控制其表達,獲得工程菌N-9和N-10。
斜面培養基:葡萄糖5 g/L,蛋白胨10 g/L,酵母粉5 g/L,牛肉膏10 g/L,NaCl 5 g/L,瓊脂20 g/L。
搖瓶種子培養基:葡萄糖20 g/L,蛋白胨3 g/L,酵母粉5 g/L,無水檸檬酸2 g/L,甲硫氨酸0.5 g/L,KH2PO42.5 g/L,MgSO4·7H2O 0.5 g/L,FeSO410 mg/L,VB1、VB3、VB5、VB12各1 mg/L,pH 7.0~7.5。
搖瓶發酵培養基:葡萄糖20 g/L,蛋白胨4 g/L,酵母粉6 g/L,無水檸檬酸2 g/L,甲硫氨酸0.3 g/L,KH2PO45.5 g/L,K2HPO45 g/L,MgSO4·7H2O 2 g/L,FeSO420 mg/L,VB1、VB3、VB5、VB12各2 mg/L,苯酚紅8 mg/L,pH 7.0~7.5。
NAM 是β-NMN 合成的重要前體,大腸桿菌中pncA編碼的煙酰胺酶可以催化NAM脫酰胺生成煙酸,是NAM 的主要支路代謝[19]。因此,首先敲除pncA基因,減少底盤細胞對NAM的額外消耗,構建出工程菌N-1。
1.1.2 培養基
引物由安升達(天津)生物科技有限公司合成。
1.1.4 試劑
蛋白胨、酵母粉 英國Oxoid公司;無水檸檬酸、甲硫氨酸、KH2PO4、K2HPO4、MgSO4·7H2O、FeSO4、(NH4)2SO4、NaCl 國藥集團試劑有限公司;NAM、β-NMN 上海阿拉丁生化科技股份有限公司;乙腈(色譜純)美國賽默飛世爾科技公司;基因組提取試劑盒、質粒提取試劑盒、膠回收試劑盒 美國Omega BioTek公司;限制性內切酶、Primer STAR HS DNA聚合酶 寶日醫生物技術(北京)有限公司;單片段快速克隆試劑盒 南京諾唯贊生物科技有限公司。
1.2 儀器與設備
PTC-1148型聚合酶鏈式反應(polymerase chain reaction,PCR)儀 美國Bio-Rad公司;5 L自動控制發酵罐 上海保興生物設備工程有限公司;U3000高效液相色譜儀、Hypersil? ODS-2 C18色譜柱 美國賽默飛世爾科技公司;SBA-40D生物傳感器分析儀 山東省科學院生物研究所。
1.3 方法
1.3.1 重組質粒構建
對于快遞包裝回收,國家有關部門制定了一個時間表。2017年,國家十部門聯合發布的《關于協同推進快遞業綠色包裝工作的指導意見》明確,到2020年,可降解的綠色包裝材料應用比例將提高到50%,基本淘汰重金屬等特殊物質超標的包裝物料,基本建成專門的快遞包裝物回收體系。主要快遞品牌協議客戶電子運單使用率達到90%以上,平均每件快遞包裝耗材減少10%以上,推廣使用中轉箱、籠車等設備,編織袋和膠帶使用量進一步減少。基本建立快遞業包裝治理體系。2019年1月1日實施的《電商法》中第五十二條規定,快遞物流服務提供者應當按照規定使用環保包裝材料,實現包裝材料的減量化和再利用。
本研究所用質粒pTrc99a自身含有表達乳糖操縱子阻遏物的lacI基因,在表達該質粒時,需添加異丙基硫代半乳糖苷(isopropyl-β-D-thiogalactoside,IPTG)進行誘導,但IPTG易影響菌體生長,因此構建組成型質粒QI-pTrc99a。通過設計兩端攜帶酶切位點ApaI的引物,利用PCR擴增出不含lacI的線性化pTrc99a載體,再通過重組酶Exnase?II的作用使其自身環化,構建出組成型質粒QI-pTrc99a,該質粒構建示意圖見圖2。根據目的基因選擇適宜的酶切位點,對質粒QI-pTrc99a進行雙酶切,利用PCR擴增獲得含酶切位點、啟動子的目的基因片段,將酶切、PCR產物回收后利用Exnase?II重組,經轉化、篩選、鑒定后獲得重組質粒。

圖2 組成型質粒QI-pTrc99a構建方法Fig.2 Flow chart for the construction of the constitutive plasmid QI-pTrc99a
1.3.2 基因組編輯
本研究利用規律成簇的間隔短回文重復/CRISPR相關蛋白9(clustered regularly interspaced short palindromic repeats/CRISPR-associated protein 9,CRISPR/Cas9)介導的基因編輯系統對菌株基因組進行改造[18]。CRISPR/Cas9基因編輯系統由pREDCas9和pGRB兩個質粒構成。溫敏型質粒pREDCas9主要由λ噬菌體的RED重組系統、Cas9蛋白表達系統和pGRB質粒消除系統組成,含奇霉素抗性(工作質量濃度100 mg/L,32 ℃)。質粒pGRB以質粒pUC18為骨架,由啟動子J23100、gRNA-Cas9蛋白結合區域和終止子組成,含氨芐青霉素抗性(工作質量濃度100 mg/L,37 ℃),該質粒是針對某一靶位點利用gRNA設計工具CRISPR RGEN Tools(http://www.rgenome.net/cas-designer/)選定gRNA序列后設計出的重組質粒。pGRB能夠轉錄出相應的gRNA,與pREDCas9表達出的Cas9蛋白結合后形成復合物并識別到靶位點,從而實現該靶位點的雙鏈DNA斷裂;此時,外源提供的根據待編輯基因設計出的重疊DNA片段(整合片段:上游同源臂-待整合基因-下游同源臂;敲除片段:上游同源臂-下游同源臂)可在重組酶作用下,與斷裂的雙鏈DNA發生同源重組,進而實現基因組改造。
通過紫外分光光度計(OD600nm)測定菌體生物量。通過SBA生物傳感儀測定葡萄糖質量濃度。通過高效液相色譜儀檢測β-NMN質量濃度,色譜條件:色譜柱為Hypersil? ODS-2 C18(4.6 mm×250 mm),流動相為95% 20 mmol/L乙酸銨∶5%乙腈(V/V),柱溫30 ℃,檢測波長254 nm,流動相總流速1 mL/min。
1.3.3 搖瓶發酵
課堂教學中教師可以利用平臺針對某個知識點或者一節課的所有知識點,做一次課堂檢測,由于平臺可以同步看到學生的答題情況,匯總統計答題的結果,這樣教師就能夠根據隨堂練習的反饋結果及時把握學生的掌握情況,及時對學生進行分層式的個性化指導,也便于后面教學內容的調整。
重組菌株通過試管斜面培養基傳代活化后,接種至30 mL種子培養基中(500 mL圓底瓶),37 ℃、200 r/min培養8~10 h后,按10%接種量轉接至30 mL發酵培養基中(500 mL擋板瓶),37 ℃、200 r/min培養24 h。發酵期間根據酸堿指示劑苯酚紅的變色情況,適當補加氨水或60 g/100 mL葡萄糖溶液。發酵培養基初始時添加0.1 g/L NAM,此后通過流加的方式保證發酵結束時共加入0.4 g/L NAM。
1.3.4 發酵罐發酵
重組菌株通過茄型瓶斜面培養基傳代活化后,接種至2 L種子培養基(5 L發酵罐)中,培養期間通過流加氨水調節發酵液pH 7.0左右,37 ℃培養,維持溶氧在20%~40%之間,通風量在2~4 L/h之間,攪拌轉速在200~700 r/min之間。當OD600nm達到10~20時,按15%的接種量接入3 L發酵培養基(5 L發酵罐)中,維持發酵液pH 7.0左右,37 ℃培養,溶氧在10%~35%之間,通風量在2~4 L/h之間,攪拌轉速400~900 r/min,發酵周期38 h。當罐內的葡萄糖耗盡時,開始以一定速率流加80 g/100 mL的葡萄糖溶液,維持罐內葡萄糖質量濃度在0.1~5 g/L之間。發酵培養基初始時添加0.5 g/L NAM,此后以一定速率流加NAM,保證在發酵結束時共加入5 g/L NAM。
將超聲技術應用于神經阻滯進程中,能促使周圍神經可視化,進而促使神經阻滯的有效率相應提升。當下,超聲技術在神經阻滯麻醉應用范疇不斷拓展,其具有二維分辨率高的優勢,多普勒效應能檢測出低血流信號,超聲技術聯合神經刺激儀進行神經阻滯,能夠協助麻醉醫生全過程均能看到針的移動情況,觀察麻醉的散布情況,有助于明顯提升阻滯神經定位的精確性,對血管、神經基本不產生影響,術前準備時間較為短暫。對于股神經,閉孔神經等位置相對較淺的神經,若單獨使用神經刺激儀,那么神經具體位置確認難度較大、阻滯成功率較低、術前準備時間較長、神經阻滯見效時間較長且并發癥發生率較高[2]。
1.3.5 檢測與分析
滑坡穩定性計算及滑坡推力計算,其目的是為滑坡在不同工況條件下的穩定性評價及滑坡防治提供設計依據。計算荷載考慮滑體自重、地表荷載、暴雨、動荷載、地震等因素。按《滑坡防治工程設計與施工技術規范》(DZ/T0219-2006),滑坡治理根據其危害對象程度及潛在經濟損失,滑坡穩定性計算工況、荷載組合及抗滑穩定安全系數見表3。
在重組菌株構建時,同時將pGRB質粒和重疊DNA片段電轉至已導入pREDCas9質粒的感受態細胞中,復蘇2 h后涂布于含100 mg/L氨芐青霉素和奇霉素的LB固體培養基上,挑選單菌落進行菌落PCR篩選陽性轉化子。將陽性轉化子轉接至含10 mmol/L阿拉伯糖和奇霉素的LB液體培養基中,誘導pREDCas9中的pGRB質粒消除系統表達,通過菌落PCR篩選已丟失pGRB的陽性菌株轉接至LB液體培養基中,42 ℃培養12 h,消除溫敏性質粒pREDCas9,最終經過菌落PCR篩選出無質粒的重組菌株。
1.4 數據處理
使用Origin 2019對數據進行處理和作圖分析,使用Adobe Photoshop 2020和Visio 2019對圖片進行處理組合和繪制。
2 結果與分析
2.1 減少底盤細胞對前體和產物的額外消耗
發酵罐發酵培養基:葡萄糖20 g/L,蛋白胨4 g/L,酵母粉6 g/L,無水檸檬酸2 g/L,甲硫氨酸0.3 g/L,KH2PO41 g/L,K2HPO45 g/L,MgSO4·7H2O 2 g/L,FeSO420 mg/L,(NH4)2SO42 g/L,VB1、VB3、VB5、VB12各2 mg/L,微量元素2 mL/L,pH 7.0~7.5。
目標產物β-NMN作為NAD+的直接前體物,在大腸桿菌中的代謝較為活躍,因此需要對其支路代謝途徑進行改造。β-NMN的支路代謝主要分為3 種(表3),一是通過pncC編碼的煙酰胺核苷酸酰胺水解酶轉化為NaMN[20];二是通過nadR編碼的煙酰胺核苷酸腺苷酰轉移酶轉化為NAD+[21];三是通過一系列的核苷酸酶降解為NR。根據文獻報道,與β-NMN降解相關核苷酸酶基因包括ushA、aphA、nagD、yjjG、surE[22-26]等。
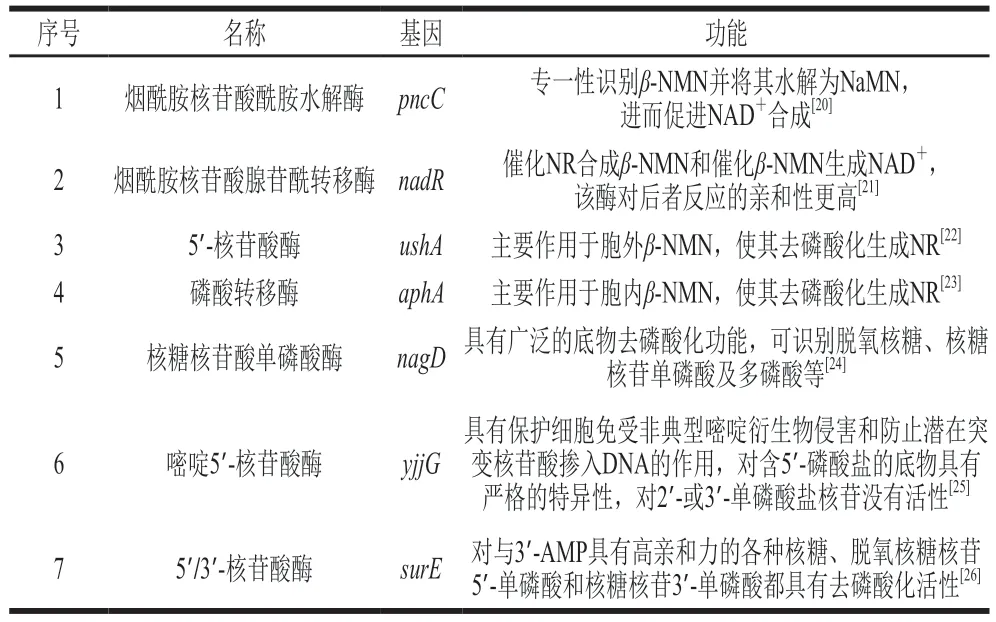
表3 大腸桿菌中β-NMN的主要支路代謝Table 3 Major β-NMN shunt metabolic pathways in E.coli
在工程菌N-1的基礎上,對β-NMN支路代謝途徑中的酶進行逐一敲除,獲得工程菌N-2~N-8。將上述菌株放到含有0.5 g/Lβ-NMN的搖瓶發酵培養基中培養16 h,考察改造后細胞對產物的消耗情況,結果如圖3所示。敲除pncC和nadR基因并不會減少細胞對β-NMN的降解,說明β-NMN向NaMN和NAD+的支路代謝并不活躍。敲除ushA后,底盤細胞對β-NMN的消耗量從100%顯著降低至25.16%,說明大腸桿菌內β-NMN向NR的轉化是β-NMN的主要支路代謝。繼續敲除其他核苷酸酶基因aphA、nagD、yjjG和surE后,底盤細胞對β-NMN的消耗量持續降低。最終,工程菌N-8對產物β-NMN的消耗量僅為4.57%,生物量與N-1相比下降了8.84%。通過上述一系列的基因改造,獲得了能夠用于β-NMN積累的優良底盤。
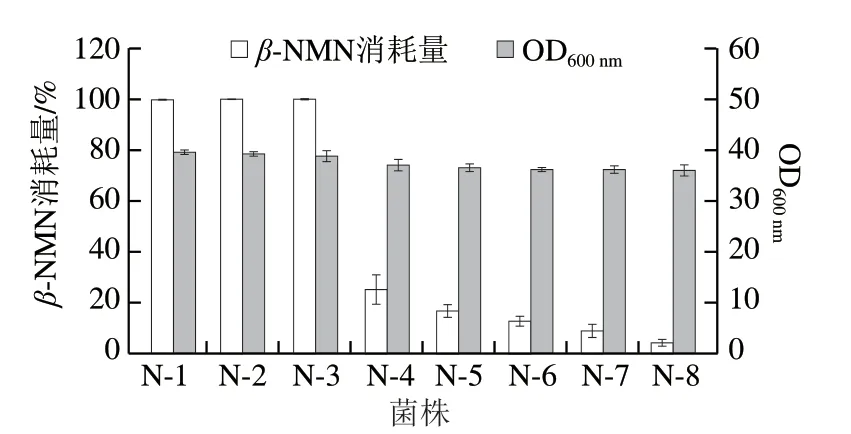
圖3 底盤細胞改造對產物β-NMN消耗以及菌株生長的影響Fig.3 Effect of chassis cell modification on β-NMN consumption and cell growth
2.2 構建β-NMN的合成代謝途徑
LB培養基:蛋白胨10 g/L,酵母粉5 g/L,NaCl 10 g/L。
彈幕(Barrage)顧名思義是子彈多而形成的幕布,在軍事上是指大量密集的炮火攻擊。在彈幕視頻中指的是大量的評論在屏幕上劃過,像是幕布一樣。最早也是最著名的彈幕網站是日本的Niconico動畫網站,其最大特點是實時彈幕發送,發送后3s就顯示在屏幕上,使得觀眾有一起參與的感覺,取得了空前成功。國內先引進彈幕技術的是Acfun彈幕視頻網和Bilibili動漫網站,隨后主流視頻網站如土豆、愛奇藝和優酷等也相繼引入彈幕技術。這種實時交流,產生共鳴的體驗感,越來越受到各個領域的廣泛關注。
PRPP是β-NMN合成的重要前體,主要是由Prs催化ATP上的焦磷酸基轉移到5-磷酸核糖的1-羥基位而產生[28]。為了強化大腸桿菌中PRPP的合成代謝,首先將內源的prsA基因整合至工程菌N-10基因組gapC位點上,并使用其內源Pprs啟動子控制其表達,獲得工程菌N-11。調節基因purR對大腸桿菌嘌呤生物合成途徑所有結構基因的表達起著負調控作用[29]。因此本研究敲除工程菌N-11的purR基因以促進prsA基因的表達,獲得工程菌N-12。
在上述代謝改造的基礎上,將使用組成型質粒(1.3.1節)表達Chitinphaga pinensis來源的Nampt,保證關鍵酶的拷貝數。將重組質粒QI-pTrc99a-namptCp分別轉化至N-9~N-12菌株,獲得工程菌N-9’~N-12’,并對其進行搖瓶發酵測試。發酵過程中添加0.5 g/L前體NAM,發酵周期24 h,結果如圖4所示。引入BcNiaP菌株N-9’的發酵液中只檢測到0.01 g/L的β-NMN,而引入BmPnuC后,菌株N-10’能夠產生0.16 g/L的β-NMN,說明大腸桿菌自身并不存在β-NMN外排系統,必須依賴外源轉運蛋白BmPnuC才能實現產物的胞外積累。強化prs基因表達以及敲除purR基因能夠使β-NMN的產量提高1.1 倍(0.34 g/L),說明上述基因改造能夠有效提高PRPP供應,為β-NMN合成提供更充足的前體。但是,prs基因過表達會導致生物量下降7.8%。
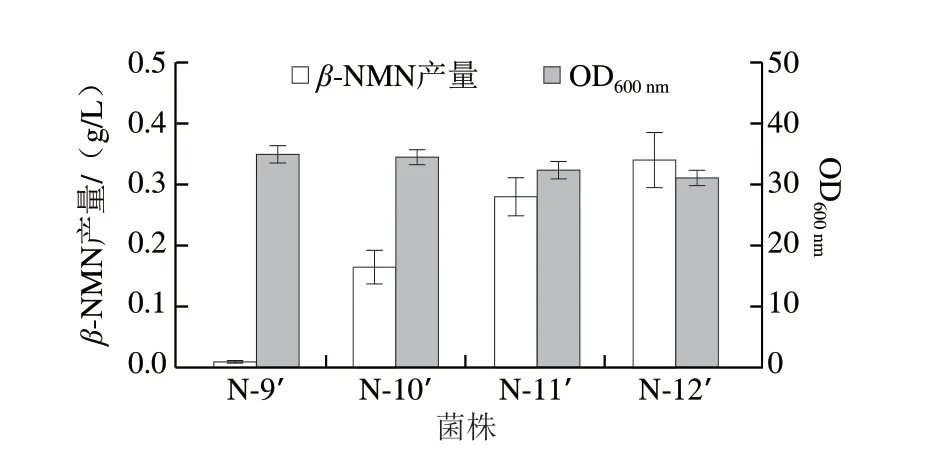
圖4 β-NMN的合成代謝途徑構建對菌株生長和β-NMN發酵的影響Fig.4 Effect of the construction of β-NMN anabolic pathway on cell growth and β-NMN fermentation
2.3 篩選高活性的Nampt
Nampt是β-NMN合成的限速酶,篩選高活性的Nampt是提升β-NMN發酵產量的關鍵。根據文獻報道,挑選5 個能夠在大腸桿菌中表達的Nampt,分別來自于Chitinphaga pinensis、Comamonadaceae bacterium、Meiothermus ruber、Haemophilus ducreyi和Vibriobacteriophage KVP40[30-31]。將不同來源的nampt基因分別與組成型質粒(1.3.1節)QI-pTrc99a連接,構建出重組質粒轉化至N-12菌株,獲得工程菌N-12′~N-16,并對其進行搖瓶發酵測試。發酵過程中添加0.5 g/L前體 NAM,發酵周期24 h,結果如圖5所示。引入VpNampt菌株N-16的β-NMN產量最高(0.90 g/L),但生物量最低,這可能與該酶是噬菌體來源有關。與之相比,引入CbNampt菌株N-13的β-NMN產量達到0.88 g/L,但生物量與N-16相比提高了1.3 倍。廖一波[32]利用分子對接手段,測定了不同來源Nampt與NAM的對接效果,發現CbNampt和MrNampt是細菌來源中評分最高的,但CbNampt的kcat/km值更高,達到9.86×106s-1。研究進一步證實,Comamonadaceae bacterium來源的Nampt在大腸桿菌中的作用效果最好,能夠在保證細胞生長的同時提升β-NMN的發酵產量。

圖5 不同來源Nampt對菌株生長和β-NMN發酵的影響Fig.5 Effects of Nampts from different sources on cell growth and β-NMN fermentation
2.4 加強PRPP供應及產物外排對β-NMN的影響
根據化學計量方程,在搖瓶發酵中添加0.5 g/L前體NAM,理論上能夠獲得1.37 g/L的β-NMN。但N-13菌株搖瓶發酵只產生了0.88 g/L的β-NMN,為理論轉化率的64.2%。菌株N-13中關鍵酶Nampt是以質粒形式表達的,而Prs和BmPnuC是以基因組整合方式表達,因此推測可能是前體PRPP供應或產物外排不足限制了β-NMN的合成。在質粒Z-2的基礎上,構建了namptCb-prsA雙基因串聯質粒Z-6,以及namptCb-prsA-pnuCBm三基因串聯質粒Z-7,并將其分別轉化至N-12菌株,獲得工程菌N-17和N-18。對上述菌株進行搖瓶發酵測試,發酵過程中添加0.5 g/L前體 NAM,發酵周期24 h,結果如圖6所示。強化prsA基因表達后,菌株N-17的β-NMN產量比N-13提高了44.3%(1.27 g/L),但生物量下降了55.1%。強化pnuCBm基因表達后,菌株N-18的β-NMN產量進一步提高,達到1.36 g/L,接近理論轉化率,但生物量比N-13下降了48.7%。發酵結果表明,加強PRPP供應及產物外排能夠有效提高β-NMN的發酵產量,但相關基因的表達強度需要嚴格控制,減少其對細胞生長的負面影響。

圖6 加強PRPP供應及產物外排對菌株生長和β-NMN發酵的影響Fig.6 Effect of strengthening of PRPP supply and product efflux on cell growth and β-NMN fermentation
2.5 工程菌N-18分批補料發酵生產β-NMN
為了測試菌株N-18的發酵性能,在5 L發酵罐中進行分批補料發酵實驗。發酵過程中添加5 g/L前體NAM,結果如圖7所示。4~26 h,隨著菌體生長,β-NMN產量不斷增加,并保持較高的合成速率。當菌體生長進入穩定期后,隨著發酵時間延長,菌體的攝糖速率變慢,β-NMN的合成速率有所降低。發酵36 h,β-NMN產量達到10.2 g/L,NAM到β-NMN的摩爾轉化率為74.5%。此外,發酵液中存在1.2 g/L的副產物煙酸,說明除了PncA以外,大腸桿菌內可能存在未知的煙酰胺酶。

圖7 工程菌N-18在5 L發酵罐分批補料發酵曲線Fig.7 Time course of fed-batch fermentation of the engineered strain N-18 in a 5 L bioreactor
目前,已有一些發酵法生產β-NMN的報道,發酵方式主要為全細胞催化或者流加NAM發酵,但都需要在產酶或者發酵階段添加誘導劑(表4)。Huang Zhongshi等[31]證明噬菌體來源的VpNampt效果最好,強化PRPP供給和產物外排后,工程菌能夠發酵產生16.2 g/L的β-NMN,NAM到β-NMN的摩爾轉化率為97%,均為已有報道的最高水平。對比了不同來源的Nampt,發現細菌來源的CbNampt效果與VpNampt相差不大,且對菌體的生長負擔較小。在5 L發酵罐補料發酵過程中,無需添加誘導劑即可實現β-NMN發酵生產,菌體生物量較高。但是,由于副產物煙酸的產生,限制了底物向β-NMN的轉化。值得注意的是,在搖瓶發酵中并沒有檢測到煙酸積累,煙酰胺幾乎完全轉化為β-NMN。但在分批補料發酵過程中,當菌體濃度較高時,煙酸開始形成,β-NMN生產速率顯著下降。根據上述現象,后續可以從代謝工程和發酵工程角度提升工程菌的發酵水平:一是通過啟動子和RBS序列優化Nampt、Prs和BmPnuC的表達水平,并利用生物信息學篩選未知的煙酰胺酶;二是調整底物流加方式和流加時間,防止過量補加NAM;三是調整補糖速率和溶氧水平,防止菌體的過度生長,減少副產物的產生。
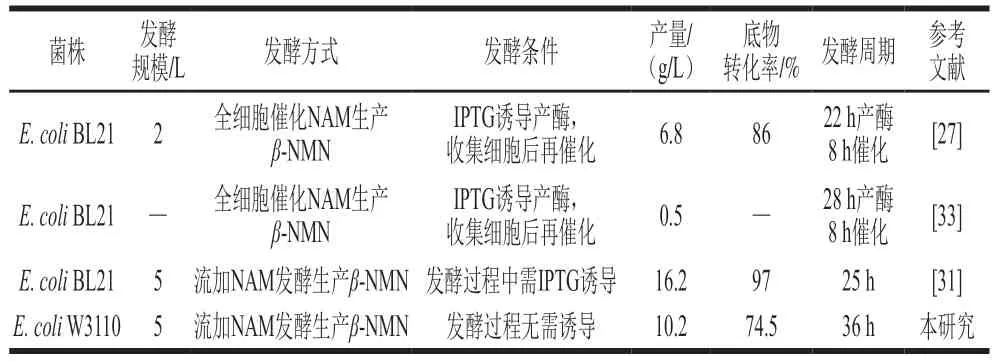
表4 不同β-NMN發酵方法對比Table 4 Comparison of β-NMN production using different fermentation methods
3 結論
研究以野生型大腸桿菌為底盤細胞,通過系統代謝工程改造,實現了β-NMN的發酵生產。主要策略包括:1)對NAM和β-NMN的支路代謝涉及的8 個酶進行失活,減少底盤細胞對前體和產物的額外消耗;2)引入外源NAM和β-NMN的轉運蛋白以及Nampt,增強胞內PRPP供給,實現了β-NMN的初步積累(0.34 g/L);3)比較篩選了酶活力較高且對底盤細胞生長負擔較小的Nampt,通過進一步優化PRPP供應和產物外排,β-NMN的搖瓶發酵產量提高至1.36 g/L。工程菌N-18于5 L發酵罐中發酵38 h,β-NMN產量可達10.2 g/L,NAM到β-NMN的摩爾轉化率為74.5%。研究構建的β-NMN發酵菌株具有遺傳背景清晰、無營養缺陷、無需誘導等優勢,可為β-NMN的工業化發酵生產及菌株改良提供理論依據。
