單鏈交聯納米粒子的合成及流變學研究
2023-12-06 12:09:24張昊巖劉庚鑫
山東化工 2023年20期
張昊巖,劉庚鑫
(東華大學 材料科學與工程學院 先進低維材料中心化纖與高分子材料改性國家重點實驗室,上海 201620)
對于各種納米粒子的研究一直是材料及化學領域的重要方向,為醫學、航天、電子等多個領域帶來了希望和挑戰。單鏈交聯納米粒子(Single-chain cross-linked nanoparticles,簡稱SCNP),是一種由單鏈高分子進行鏈內折疊及交聯反應得到的納米粒子,具有許多獨特的性能,吸引了多個領域的科學家對其進行研究,例如在納米醫學中有廣泛的應用,可作為催化納米反應器,傳感器和藥物遞送的載體。近些年,高分子合成化學的發展使得合成結構復雜、分子量分布窄、單體和官能團序列可控的聚合物成為可能。為此,人們開發了不同的合成策略來設計單鏈納米粒子。聚合物分子內交聯所選擇的特殊合成方法會影響SCNP的結構,從而影響其性能[1]。早在1956年,Kuhu 和Majer[2]就報道了線性高分子鏈通過分子內交聯反應形成SCNP的概念。直到今天,關于SCNP的合成一直是研究的熱門課題。關于SCNP的有兩項關鍵的技術:(1) SCNP前驅體鏈的精確、可控合成;(2)通過各種共價、非共價鍵實現分子內交聯[3]。對于前驅體鏈的合成,原子轉移自由基聚合是一種較為廣泛使用的方法。原子轉移自由基聚合(Atom transfer radical polymerization,簡稱ATRP)是一種由過渡金屬參與的可控-活性自由基聚合,目前已被廣泛使用。自發明以來就在1990年代中期到末期[4-7]合成了各種各樣的功能性聚合物的同時控制分子量和分散度。
對于分子內交聯的方法,有環加成反應,光引發反應,銅催化的“點擊”化學反應,邁克爾加成等等。苯并環丁烯反應是分子內交聯反應中最具影響力的化學反應之一[8]。它的主要機理是苯并環丁烯基團開環,形成極活性的鄰醌結構,主要通過不可逆二聚反應生成二苯并環辛二烯衍生物,以及一種未鑒定的低聚物混合物。這種方法通過使用連續加成方法結合熱激活苯并環丁烯偶聯化學,即使在較高濃度(0.1 mol/L)下也可以有效地消除分子間的交聯,這使其成為大規模合成結構明確的納米粒子的實用技術。因此,我們可以利用此反應來合成SCNP。
流變學這門學科研究的是材料的流動與變形。遵循牛頓定律的流體被叫做牛頓流體,遵從胡克定律的材料被叫做胡克彈性體[9]。然而在現實生活中,物質的流動伴隨著形變,牛頓流體和胡克彈性體是兩類被簡化的理想對象。然而,真實的材料其流動變形遠比想象的要復雜。如:食品,血漿,蛋清,橡膠,纖維紡絲液等等。它們在流動時黏性與彈性均存在,因此,在流動變形過程中伴隨著彈性記憶效應,那么這種既有黏性又有彈性的性質就是流變學需要研究的特有的行為。單純地引用牛頓流動定律或者胡克定律不能很好地描述這類材料的流動和變形。而流變學研究的主要是非牛頓流體,即用胡克彈性模型和牛頓流體理論都不能準確描述其復雜力學特性的材料。聚合物流體就是典型的非牛頓流體,通過流變實驗和分析研究,人們可以獲取很多關于聚合物的性質:聚合物的分子量和分子量分布,聚合物的黏彈性等。
本研究主要是合成一種基于乙烯基苯并環丁烯的單鏈交聯納米粒子,并對其流變學行為進行研究。首先是通過ATRP的方法合成一系列以苯乙烯與4-乙烯基苯并環丁烯為單體的無規共聚物,之后將該無規共聚物作為前驅體通過連續滴加法合成SCNP。得到的產物進行核磁共振譜、凝膠滲透色譜、動態光散射、差示量熱掃描等表征。同時我們使用商用流變儀進行小幅振蕩剪切測量,通過測得的主曲線可得到松弛時間及黏度,以研究SCNP及其前驅體的流變學行為。
1 實驗材料及方法
1.1 實驗試劑
本實驗使用的試劑藥品如下:苯乙烯(St),二芐醚(DBE)購自上海阿拉丁生化科技有限公司,其中苯乙烯經中性氧化鋁柱純化去除阻聚劑,二芐醚經過凍抽循環三次除氧。不含該阻聚劑的4-乙烯基苯并環丁烯(BCB)購自畢得醫藥。2-溴異丁酸乙酯(Ebib),五甲基二乙烯基三胺(PMDETA),溴化亞銅(CuBr),甲醇購自上海泰坦科技有限公司,甲苯和四氫呋喃(THF)購自國藥控股化學試劑有限責任公司,均未經處理直接使用。
1.2 實驗設備
400 MHz核磁共振氫譜,德國布魯克公司;ACQUITY APC系統(APC),Waters公司;Zetasizer Nano ZS粒徑儀,馬爾文公司;DSC250型差示掃描量熱儀,TA公司。
1.3 實驗方法
1.3.1 前驅體鏈的合成
本課題設計的前驅體鏈合成路線如圖1所示。
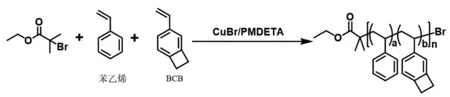
圖1 線形前驅體合成示意圖
如圖所示,我們以2-溴異丁酸乙酯為引發劑,將苯乙烯及功能性單體4-乙烯基苯并環丁烯進行無規共聚,聚合方法為ATRP。由于單體及引發劑的特性,本次聚合須在90 ℃下反應較長時間。
典型的聚合過程如下:將單體苯乙烯(2 g)、BCB(0.75 mg)、引發劑2-溴異丁酸乙酯(1.2 mg)、PMDETA (1.6 mg)溶于甲苯(1.0 mL)中,在氮氣流量恒定的條件下,加入催化劑溴化亞銅(2.0 mg)后再進行凍抽循環三次,之后將反應瓶在90 ℃的油浴中浸泡14 h。然后用THF稀釋反應混合物,將稀釋后的反應混合物通過中性氧化鋁柱去除銅離子。用旋轉蒸發法去除溶劑,用甲醇三次沉淀制得共聚物PS-389k[10]。
1.3.2 單鏈交聯納米粒子的合成
前驅體鏈在進行分子內交聯合成SCNP時,使用了連續滴加法,即在反應體系的濃度接近極稀溶液的狀態來抑制分子間交聯反應。該反應體系須整個達到250 ℃進行才可合成SCNP。與前驅體鏈類似,數均、重均分子量,聚合物多分散性指數均由GPC得出,流體力學直徑Dh由DLS測得。具體數據如表1所示。

表1 樣品分子量及粒徑總結表
具體步驟如下:
在250 mL的三頸燒瓶中,將60 mL DBE在氮氣下加熱到250 ℃,并將溶解在10 mL DBE中的共聚物(0.15 g)以約4.8 mL/h的速度通過蠕動泵滴入,并在氮氣氛圍下強力攪拌。溶劑加入后靜置1 h,沉淀到150 mL甲醇中。將混合物離心去除上清液,得到淡黃色固體,將軟納米顆粒在甲醇中沉淀三次,得到SCNP-389k。
1.3.3 表征
使用核磁共振氫譜(1H NMR)能間接表現出樣品的化學結構,從而可以計算出前驅體中BCB的含量。測試的具體過程如下:取8 mg左右的樣品溶于0.8 mL氘代氯仿,之后將溶液裝入核磁管中,再將該核磁管放入核磁共振譜儀中于25 ℃的溫度下進行測試。
凝膠滲透色譜(GPC),有時也被叫作體積排除色譜(SEC),我們根據其流出時間得出的曲線并與標準樣品進行比較即可得到樣品的分子量及其分布。 測試的具體過程如下:流動相為四氫呋喃(色譜級),柱溫為35 ℃,探測器溫度為35 ℃,設置流速為1 mL/min。取3 mg樣品溶于四氫呋喃(色譜級)中,靜置12 h,待其完全溶解后(溶液澄清透明為標準)用0.22 μm孔徑的聚四氟乙烯過濾頭過濾,之后使用注射器將溶液注射到儀器中進行測試。
動態光散射(DLS)主要是根據不同粒徑大小的微粒產生的散射光強不同來得到不同的散射強度的變化曲線,從而得出粒徑分布規律。將樣品配成1 mg/mL濃度的溶液,然后使用孔徑為0.22 μm的聚四氟乙烯濾頭過濾,之后注入到石英比色皿中。將石英比色皿放入儀器的測試架,測試前需保持環境溫度在25 ℃,平衡2 min。測試重復15次,所得函數曲線通過算法計算平均粒徑和多分散系數以及粒徑分布等。
差示掃描量熱 (DSC) 測試指在相同的溫度變化下,用補償器測量樣品與參比物之間的溫差保持為零所需熱量對溫度T的依賴關系,從測到的曲線我們可以算出玻璃化轉變溫度(Tg)等數據。儀器采用TA公司的DSC250。在氮氣氣氛下保護下設置溫度在30 ~ 180 ℃的范圍內對樣品進行10 ℃/min的升溫-降溫過程,再以10 ℃/min進行升溫,在第二次的升溫曲線上讀取Tg來減少熱歷史造成的誤差。
1.3.4 流變測量
具體步驟以SCNP-389k為例:
取50 mg樣品,將其制成8 mm的薄片。將儀器升至120 ℃預熱后調整上下夾具的間隙至0。將儀器溫度升至Tg以上約20 ℃,待溫度穩定后上樣,使傳感器和樣品充分接觸,并刮去多余樣品。之后設定好儀器測試程序即可開始測試。測試采用小幅振蕩剪切測試(Small oscillating shear,簡稱SAOS)模式,測試頻率范圍在0.01~100 rad/s,在約120 ℃ 至160 ℃范圍內的不同溫度下進行。測量得到的曲線經過時間-溫度疊加(Time-temperature superposition,TTS)處理得到主曲線,為減小誤差參考溫度選擇為Tref=160 ℃。
2 結果與討論
2.1 SCNP的核磁共振氫譜(1H NMR)
圖2顯示了前驅體鏈PS-389k的結構圖,圖3則是前驅體鏈PS-389k和單鏈交聯納米粒子SCNP-389k的核磁共振氫譜。由于苯乙烯和4-乙烯基苯并環丁烯兩種單體的主要化學結構差別較小,因此前驅體的氫所處化學環境相近,核磁共振氫譜的化學位移也相似。更加明顯的特征峰是4-乙烯基苯并環丁烯的四元環,化學位移為3.04,如圖3所示,與之前的研究結果類似[10]。在形成SCNP后由于苯并環丁烯的四元環會與臨近的相同結構兩兩反應形成新的八元環,因此如圖3所示,SCNP 3.04處的特征峰會消失,取而代之的是在2.0~3.0范圍內觀察到寬共振峰,由此證明分子內交聯反應比較完全。由于四種交聯劑含量的前驅體鏈中氫的化學環境一致,因此我們可以通過圖中b,c兩處峰面積的比值可計算交聯劑的含量即交聯度,代入公式為。

圖2 前驅體鏈PS-389k結構示意圖

圖3 前驅體鏈及SCNP核磁共振氫譜圖
(1)
式中x為BCB的含量,最后算出交聯劑的含量為8%。
2.2 SCNP的凝膠滲透色譜(GPC)
圖4為SCNP-389k及其前驅體PS-389k的GPC流出時間對比圖。在圖中我們可以看到,與前驅體相比,SCNP分布曲線明顯向更長的流出時間移動,表明交聯后SCNP的流體動力學半徑明顯減小,即SCNP的尺寸明顯小于相應前驅體鏈的尺寸,這與以往的研究相符[10]。我們還發現SCNP的流出時間曲線分布明顯較前驅體的流出時間曲線略寬,結合PDI測試結果我們認為這種大分子量的高分子鏈在溶液中尺寸較大,會不可避免地產生耦合的情況,導致生成二聚體或多聚體等。

圖4 凝膠滲透色譜(GPC)流出時間分布曲線
2.3 SCNP的動態光散射(DLS)
圖5是SCNP和前驅體鏈的粒徑分布圖。我們可以從圖中看出, SCNP-389k與前軀體鏈PS-389k相比,流體力學直徑明顯降低,說明其經過分子內交聯反應后分子結構進行塌縮,產生了一種較為緊密的構象。我們也發現了SCNP粒徑分布曲線拓寬的情況,推測也是由于交聯過程中不可避免的耦合導致的。

粒徑/nm
2.4 SCNP的差示掃描量熱 (DSC)
圖6為SCNP及前驅體的DSC變溫曲線。從圖中我們可以看出,相較于前驅體鏈,SCNP的玻璃化轉變溫度(Tg)明顯升高,同時其玻璃化轉變的范圍變寬。造成這種現象的主要原因是SCNP內部的交聯網絡限制了鏈段的運動,從而導致其Tg上升。
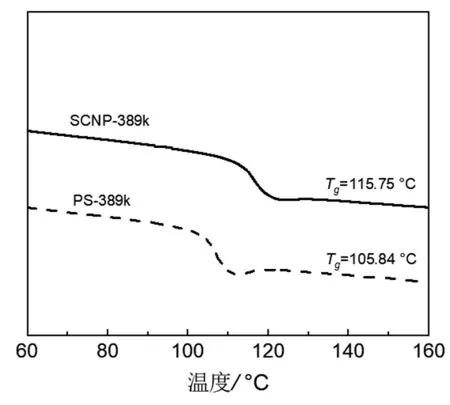
圖6 前驅體鏈及SCNP的差示掃描量熱(DSC)升溫曲線
2.5 流變測試
如圖7所示為SCNP及其前驅體的SAOS曲線。根據以往的研究我們可以知道SCNP樣品的末端松弛時間可以用儲能模量G′和 損耗模量G′′ 的交點頻率ωco的倒數表示,即τ=ωco。因此,對于前驅體鏈PS-389k來說,其松弛時間。然而,在SCNP-389k的SAOS曲線末端并沒有出現交點,因此我們計算需要將末端G′、G′′的近似直線區域擬合直線并反向延長,交點橫坐標倒數即τ。我們通過計算得出其松弛時間τ=1.4 s,相較于前驅體略低,說明SCNP的熔體動力學相較于前驅體會變快。

圖7 前驅體及SCNP樣品的小幅振蕩剪切(SAOS)主曲線
如圖8所示為前驅體及SCNP的復數黏度(η*)與頻率的函數關系圖。我們可以看出,在頻率趨近于0時,SCNP的黏度明顯小于前驅體鏈的黏度,說明分子內交聯反應后的聚合物零切黏度有明顯的降低趨勢。
圖8 前驅體及SCNP樣品的復數黏度(η*)主曲線
3 結論
合成了一種基于乙烯基苯并環丁烯的單鏈交聯納米粒子,通過核磁共振譜、凝膠滲透色譜、動態光散射、示差量熱掃描等方法表征,最后將樣品進行小幅振蕩剪切模式的流變測試。本論文的具體結論如下:
(1)單鏈交聯納米粒子交聯后尺寸明顯減小。相較于前驅體,SCNP的分子內部為較為緊密的構象。
(2)單鏈交聯納米粒子獨特的拓撲結構能夠使熔體的松弛變快,從而使其黏度降低。根據這種特點SCNP可作為填料降低材料的黏度,提高材料的可加工性。
