高效液相色譜-串聯(lián)質譜法同時測定水產品中10 種硝基咪唑類藥物殘留量
2023-11-07 06:26:56謝云波童文羽朱志強石義付
湖北農業(yè)科學 2023年10期
關鍵詞:檢測
謝云波,易 鳴,童文羽,曾 智,朱志強,石義付
(湖北省水產科學研究所,武漢 430071)
硝基咪唑(NMZs)類藥物是一類合成的人和動物常用的廣譜抗菌和抗原蟲藥物,在水產養(yǎng)殖中常被用于防治和治療寄生蟲感染[1],常見的有甲硝唑、洛硝噠唑和地美硝唑等。但由于硝基咪唑類藥物含有的硝基雜環(huán)具有細胞誘變性、致癌性和潛在的致畸性,經生物體代謝后產生的帶有側鏈羥甲基化合物具有與原藥相似的毒性,其殘留對水產品質量安全構成了直接威脅[2],許多國家和地區(qū)已禁止該類藥物用于食源性動物,中國也于2002 年將該類藥物列入《食品動物禁用的獸藥及其它化合物清單(中華人民共和國農業(yè)部公告第193 號)》[3]。
目前水產品中硝基咪唑類藥物殘留量檢測的國家標準包括GB/T 21318—2007《動物源性食品中硝基咪唑殘留量檢測方法》[4]和《動物源性食品中4 種硝基咪唑殘留檢測液相色譜-串聯(lián)質譜法(中華人民共和國農業(yè)部公告第1025 號)》[5],地方標準包括浙江省質量技術監(jiān)督局發(fā)布的DB33/T 693—2008《動物源性食品中硝基咪唑類藥物殘留量的測定高效液相色譜法》[6],其中《動物源性食品中4 種硝基咪唑殘留檢測液相色譜-串聯(lián)質譜法(中華人民共和國農業(yè)部公告第1025 號)》[5]僅能檢測甲硝唑、羥甲基甲硝咪唑、洛硝噠唑、二甲硝唑;DB33/T 693—2008《動物源性食品中硝基咪唑類藥物殘留量的測定高效液相色譜法》[6]也只能檢測甲硝唑、洛硝噠唑、地美硝唑、替硝唑、奧硝唑,且因為是液相法,靈敏度較低;GB/T 21318—2007《動物源性食品中硝基咪唑殘留量檢測方法》[4]能檢測甲硝唑、洛硝噠唑、地美硝唑、氯甲硝咪唑、苯硝咪唑、異丙硝唑、2-甲硝咪唑、4-甲硝咪唑8 種原藥和羥基甲硝唑、羥甲基甲硝咪唑2 種代謝產物,但該方法前處理操作繁瑣,對實驗室硬件和操作人員水平要求較高。而且以上國家和地方標準都是針對動物源性基質,沒有針對水產品基質進行優(yōu)化,實際試驗過程中出現(xiàn)了標準曲線線性不佳,回收率不穩(wěn)定的情況。
針對以上方法的不足,本研究建立了同時測定水產品中10 種硝基咪唑類藥物(表1)含量的高效液相色譜串聯(lián)質譜法,方法靈敏度可達1 μg/kg,線性范圍為1~150 μg/L,適用于草魚、加州鱸魚、克氏原螯蝦,該方法步驟簡單、靈敏度高,適用于大批量樣品的檢測。
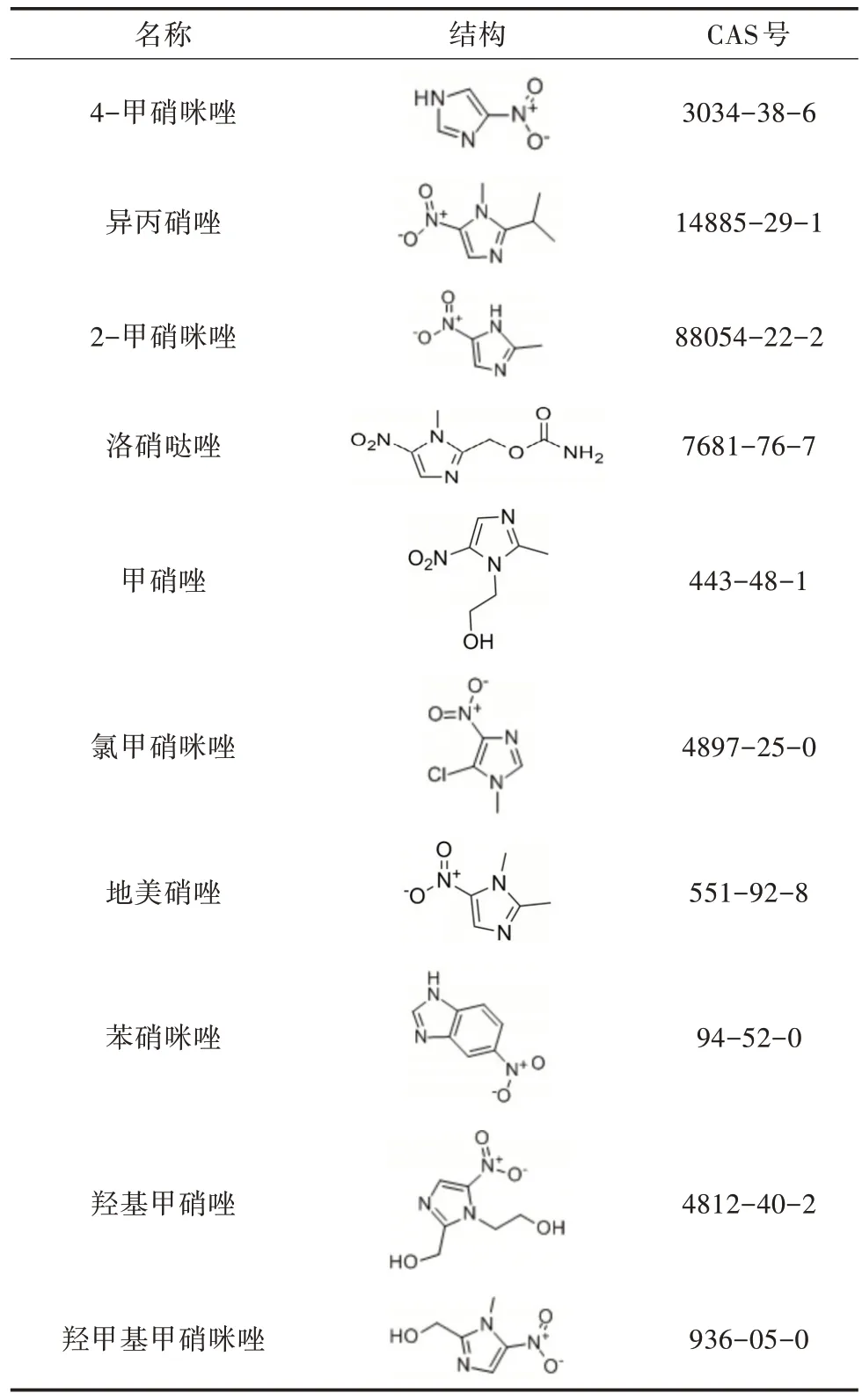
表1 10 種硝基咪唑類藥物的結構和CAS 號
1 材料與方法
1.1 材料與試劑
試驗樣品均來自湖北省白沙洲農產品大市場,品種有草魚、加州鱸魚、克氏原螯蝦。
選用10 種硝基咪唑類藥物標準品,其生產批號、純度、不確定度如表2 所示。

表2 硝基咪唑類藥物標準品信息
乙酸乙酯、氨水、正己烷均為分析純,購自國藥集團化學試劑上海有限公司;甲醇、甲酸均為質譜純,購自美國默克公司。
1.2 儀器
XPE205 型十萬分之一天平(瑞士METTLER TOLEDO 公司);RE52CS 型旋轉蒸發(fā)儀(上海亞榮生化儀器廠);KQ-500B 型超聲波儀(昆山市超聲儀器有限公司);U300 型超高效液相色譜串聯(lián)SCIEX QTRAP 4500 質譜儀(美國賽默飛公司)。
1.3 方法
1.3.1 標準溶液配制 分別稱取10 種硝基咪唑類藥物標準品10 mg,用少量甲醇溶解,超聲波助溶,置于100 mL 棕色容量瓶中定容,配制成100 mg/L 的標準儲備液,4 ℃冷藏保存,有效期3 個月。分別量取10 種硝基咪唑類藥物標準溶液,用甲醇稀釋成濃度為0.5 mg/L 的混合標準工作液,現(xiàn)配現(xiàn)用。
1.3.2 樣品制備 魚去鱗,沿背脊取可食肌肉部分,蝦去頭、殼,取可食肌肉部分,切為小塊,絞成肉糜,經GB/T 21318—2007《動物源性食品中硝基咪唑殘留量檢測方法》[4]液質法檢測確認陰性,待用。
1.3.3 樣品提取 稱取5.00 g 樣品,置于50 mL 離心管中,加入15 mL 乙酸乙酯,100 μL 氨水,加蓋后垂直振蕩(500 次/min)1 min,4 000 r/min 離心5 min,分離后取上清液,重復上述操作1 次,合并2 次上清液,40 ℃旋轉減壓蒸餾至干。
1.3.4 樣品凈化 向茄形瓶中加入2 mL 正己烷,渦旋1 min,再加1.5 mL 10%甲醇溶液,渦旋1 min,再將全部液體轉移至離心管中,4 000 r/min 離心10 s,去除上層正己烷,吸取下層溶液,過0.22 μm 微孔濾膜待上機。
1.3.5 色譜條件 進樣量:10 μL;色譜柱:島津C18(150 mm×2 mm,5 μm);柱溫:35 ℃。流動相A:0.2%甲酸水溶液;流動相B:乙腈;流速:0.4 mL/min,各組分梯度洗脫程序如表3 所示,同時為了減少離子源污染,設定進樣開始后3.5~7.0 min 流動相才進入檢測器。

表3 洗脫程序
1.3.6 儀器質譜條件 離子源:ESI;掃描方式:正離子模式;檢測方式:質譜多反應監(jiān)測(MRM);電噴霧電壓:5 500 V;霧化氣壓力:0.379 MPa;氣簾氣壓力:0.207 MPa;輔助氣壓力:0.379 MPa;離子源溫度:550 ℃,10 種硝基咪唑藥物的保留時間、定量離子對、定性離子對、碰撞能量等見表4。
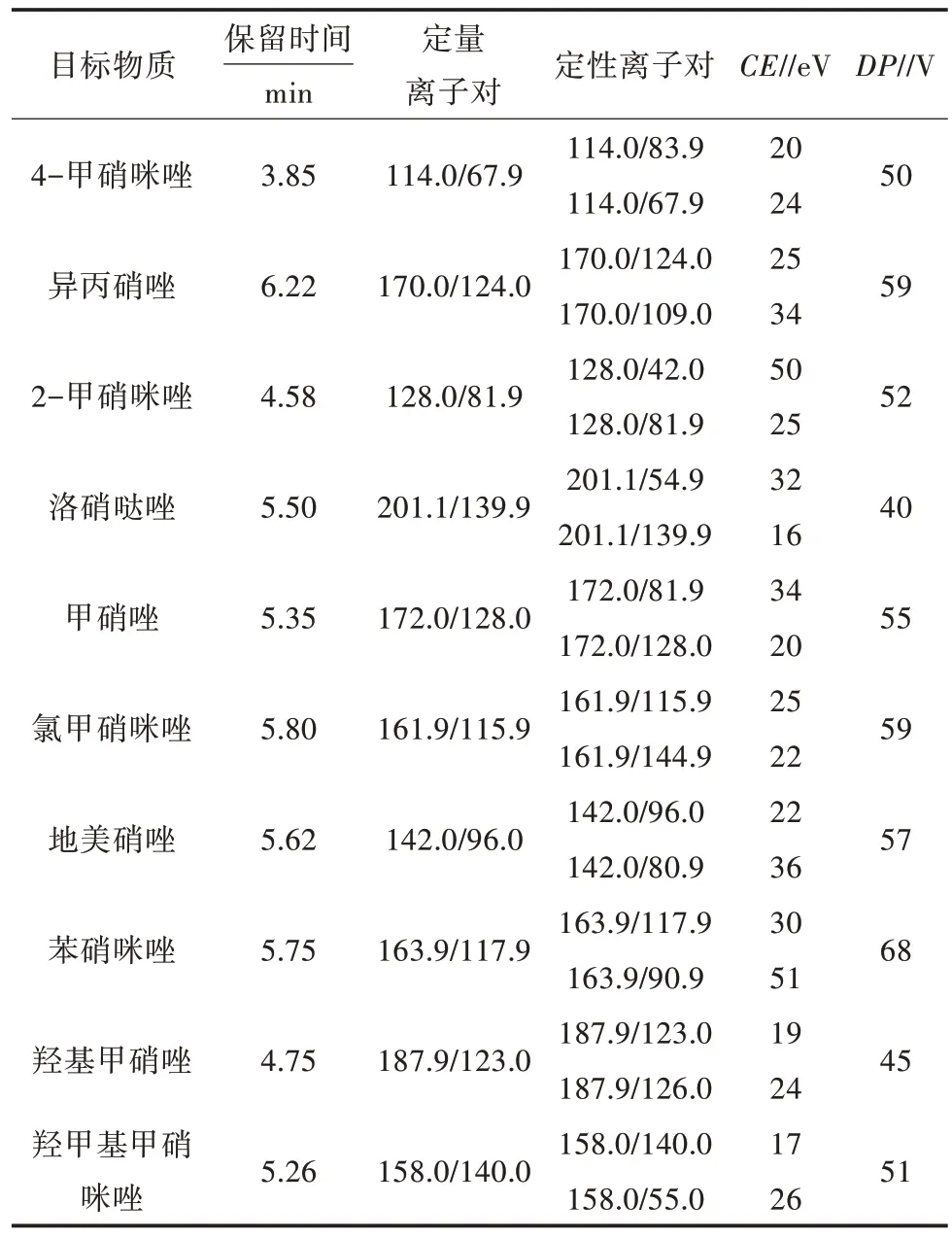
表4 10 種硝基咪唑藥物的保留時間、選擇監(jiān)測離子設定參數
2 結果與分析
2.1 前處理優(yōu)化
水產品中硝基咪唑類藥物殘留量的檢測方法主要使用乙酸乙酯、甲醇、乙腈、二氯甲烷等有機溶劑作為提取劑,如《動物源性食品中4 種硝基咪唑殘留檢測液相色譜-串聯(lián)質譜法(中華人民共和國農業(yè)部公告第1025 號)》[5]使用乙酸乙酯提取,正己烷液液萃取,MCX 固相萃取小柱凈化,HPLC-MS 檢測,外標法定量;方力等[7]用乙酸乙酯提取,經正己烷脫脂、50 mg 乙二胺-N-丙基硅烷(PSA)吸附劑吸附凈化,經0.22 μm 親水聚四氟乙烯(PTFE)濾膜過濾后直接進行上機檢測;劉艷萍[8]和王志杰等[9]使用無水硫酸鈉作為助劑,乙酸乙酯提取,HPLC-MS 檢測,內標法定量;GB/T 21318—2007《動物源性食品中硝基咪唑殘留量檢測方法》[4]使用甲醇-丙酮均質或超聲波提取,乙酸乙酯液液萃取,凝膠滲透色譜凈化,再經C18 固相萃取小柱凈化,HPLC-MS 檢測,外標法定量;DB33/T 693—2008《動物源性食品中硝基咪唑類藥物殘留量的測定高效液相色譜法》[6]用乙酸乙酯提取,正己烷液液萃取,MCX 固相萃取小柱凈化,HPLC 紫外檢測,外標法定量。以上方法中,DB33/T 693—2008《動物源性食品中硝基咪唑類藥物殘留量的測定高效液相色譜法》[6]為液相色譜法,只能檢測甲硝唑等5 種硝基咪唑,檢測限僅為2 μg/kg,靈敏度不高;GB/T 21318—2007《動物源性食品中硝基咪唑殘留量檢測方法》[4]和方力等[7]、黎翠玉等[10]的方法提取步驟較繁瑣,實際操作中出現(xiàn)了提取液易乳化、旋轉減壓蒸餾蒸不干、過柱困難、標準曲線線性差、樣品回收率低等問題;劉艷萍[8]、王志杰等[9]在乙酸乙酯中加入無水硫酸鈉作為提取助劑,但無水硫酸鈉容易吸附待測物質,影響提取效率,并且在提取劑濃縮后有鹽結晶析出,對后續(xù)的質譜檢測不利。
試驗中發(fā)現(xiàn)二氯甲烷和乙酸乙酯對硝基咪唑類藥物的提取效果較好,但二氯甲烷對人體毒性較大,不宜采用。由于硝基咪唑類藥物是兩性化合物,在弱酸性條件下以質子狀態(tài)存在,在弱堿性條件下,以游離分子狀態(tài)存在[11],因此考慮在提取劑乙酸乙酯中加入氨水。但加入的氨水太多會降低氯甲硝咪唑、洛硝噠唑的線性范圍[12]。經試驗發(fā)現(xiàn),每15 mL乙酸乙酯中加入100 μL 氨水能夠將洛硝噠唑、氯甲硝咪唑的線性范圍提高至150 μg/L。由于提取劑乙酸乙酯與水互溶性較差,因此采用離心的方式分離上清中的水與乙酸乙酯。
硝基咪唑類藥物的凈化一般有液液萃取法和固相萃取法(SPE)2 種方式。液液萃取法凈化操作相對簡單,費用也較低,但凈化效果不如固相萃取法。固相萃取法操作步驟繁瑣,回收率受操作水平和固相萃取小柱質量影響較大且串聯(lián)質譜檢測的強抗干擾能力使其對樣品的前處理凈化依賴性很低。綜上,本研究選擇加入正己烷進行液液萃取凈化。
2.2 儀器方法的優(yōu)化
在水相中加入一定比例的甲酸能促進離子化[12],考察了不同含量的甲酸水溶液(0.10%、0.15%、0.20%、0.30%)對10 種目標化合物的分離效果,結果表明,0.10%、0.15%甲酸水溶液的分離效果均不如0.20%甲酸水溶液的分離效果,0.30%甲酸水溶液與0.20%甲酸水溶液的分離效果差別不大,為保護儀器,最后選擇0.20%甲酸水溶液。流動相中加入甲醇會導致10 種硝基咪唑類藥物出現(xiàn)不同程度的峰型變寬,靈敏度下降。選擇0.20%甲酸水溶液-乙腈作為流動相,各目標化合物峰形尖銳對稱,效果最好。
2.3 結果分析
按“1.3”項方法處理加標后的樣品,10 種硝基咪唑類藥物加標樣品的離子流見圖1,加標濃度均為10.0 μg/kg;陰性樣品的離子流見圖2,線性方程、檢出限(LOD)和定量限(LOQ)見表5;分別按5.0、7.5、10.0、15.0、20.0 μg/kg 5 個加標濃度添加,每個水平測定4 次,回收率與相對標準偏差(RSD)見表6。結果表明,方法檢出限為0.04~0.30 μg/kg,定量限為0.50~1.00 μg/kg,標準曲線線性范圍為1~150 μg/L,相關系數(R2)≥0.998 00,回收率為73.0%~116.2%,相對標準偏差為0.43%~9.53%。
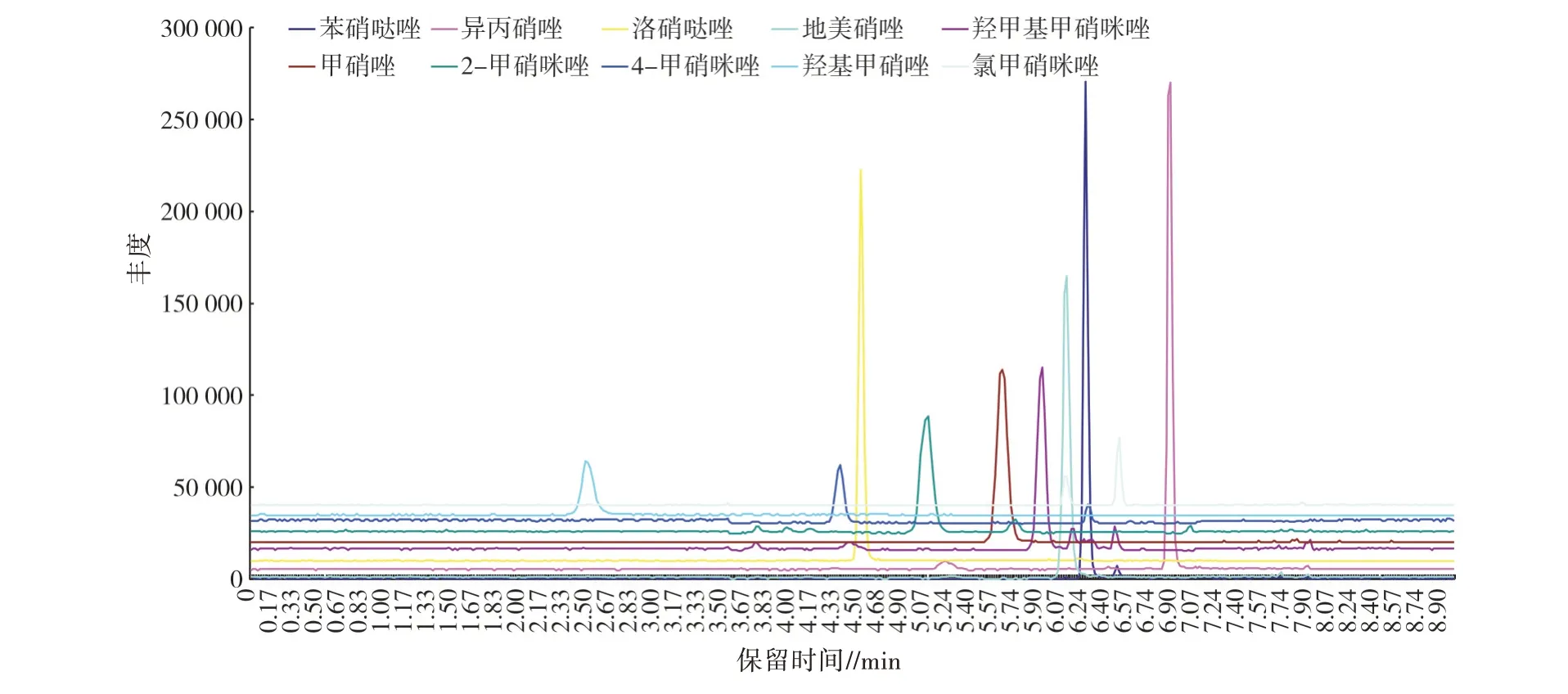
圖1 加標樣品的離子流

圖2 陰性樣品離子流
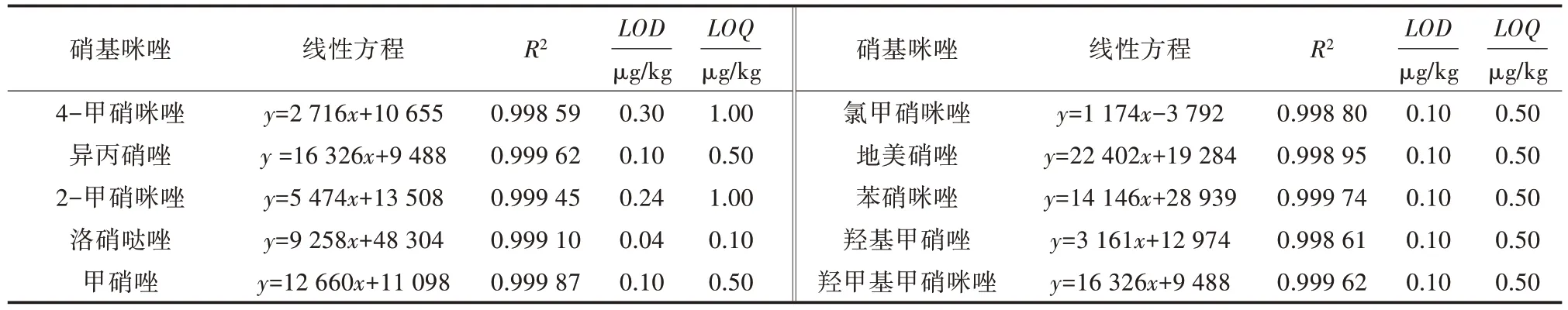
表5 10 種硝基咪唑藥物的線性方程、檢出限與定量下限

表6 方法的回收率和相對標準偏差
2.4 實際樣品檢測
利用本研究建立的方法對湖北省市售的4 批次共210 個水產品中10 種硝基咪唑類藥物進行檢測,結果發(fā)現(xiàn),10 種硝基咪唑類藥物在4 批次水產品中均未檢出。
3 小結
本研究建立了同時測定水產品中10 種硝基咪唑類藥物殘留的高效液相色譜串聯(lián)質譜法。標準曲線線性范圍為1~150 μg/L,相關系數(R2)>0.998 00,在草魚、加州鱸魚、克氏原螯蝦基質中分別添加5.0、7.5、10.0、15.0、20.0 μg/kg 硝基瞇唑藥物,回收率為73.0%~116.2%,相對標準偏差為0.43%~9.53%。該方法步驟簡單、靈敏度高、重現(xiàn)性好,適合于大批量樣品的檢測。
猜你喜歡
中國設備工程(2022年12期)2022-07-11 04:33:00
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:36
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:34
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:50
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:48
