原位電離質譜快速篩查豬肉中10種喹諾酮
2023-11-01 07:09:22馮琳琳郝莉花郭思嘉衛潤鑫楊龍松
安徽農業科學 2023年20期
馮琳琳,郝莉花*,郭思嘉,鞏 凡,衛潤鑫,楊龍松
(1.河南省產品質量檢驗技術研究院,河南鄭州 450000;2.河南省食品安全數據智能重點實驗室,河南鄭州 450000)
喹諾酮類抗生素是通過抑制 DNA 旋轉酶活性殺滅細菌的一類人工合成化合物,常見的喹諾酮類抗生素主要有恩諾沙星、氧氟沙星、培氟沙星、諾氟沙星、沙拉沙星等。喹諾酮類藥物以其抗菌譜廣、吸收好、價格低廉等特點,在臨床方面得到了較廣泛的應用。但是不合理的用藥可導致喹諾酮類藥物及其代謝產物在動物機體組織中的殘留。因此人食用動物組織后抗生素在人體內會殘留、蓄積,造成人體對該藥物的耐藥性,破壞腸道菌群,影響人體免疫系統[1],具有潛在的致癌、致突變的作用[1-2]。所以,世界各國都對該類藥物在畜禽肉中的殘留制定了嚴格的限量。因此,對于動物源性食品中喹諾酮類藥物檢測顯得尤為重要。
目前檢測喹諾酮類抗生素比較常用的方法有高效液相色譜法 (high performance liquid chromatography,HPLC)[3-4]、液相色譜-質譜法 (liquid chromatography mass spectrometry,LC-MS)[5]、微生物法(microbial inhibitions test,MIT)[6]等。HPLC 和 LC-MS 法結合了色譜的分離能力和質譜的定性能力,可對復雜化合物進行更精準、簡便的定性和定量分析,靈敏度比較高[7],但樣品前處理、操作過程較為煩瑣,檢測時間長,對試驗操作人員技術要求較高。MIT 法可操作性非常強、成本較低,但靈敏度低、敏感性差,易受其他抗生素干擾[6,8]。這些方法存在前處理復雜、操作技術性強、檢測成本高等缺陷[9]。因此,開發快速、簡便的喹諾酮類抗生素篩查方法具有較好的應用價值。
目前,藥物殘留快速篩查方法主要有膠體金免疫層析技術[10]、傳感器[11]、表面增強拉曼光譜法[12]、原位直接電離質譜等。這些方法靈敏度高、操作簡便、反應時間短,可實現藥物殘留的現場快速定量檢測,但易受人為因素干擾導致假陽性或假陰性,且不適合多種待測物的高通量快速檢測。直接離子化質譜或原位直接電離質譜,可在大氣壓環境中實現原位、直接和快速的樣品分析,不需要復雜的樣品制備過程,并可節大量的資源和時間[13-14]。在各種原位質譜電離源中,實時直接分析(direct analysis in real time,DART)離子源是一種非表面接觸的熱解析大氣壓電離技術,結合了等離子體技術的簡單性和靈敏性,可直接分析固體、液體和氣體[15-19]。DART離子源具有高檢測速度和強大的抗干擾能力,可高通量和直接分析復雜基質樣品的特性[17],DART包含了大氣壓電離源類的軟電離特性,與傳統離子源(如電子轟擊電離等)以及電噴霧電離源相比,不僅具有離子化效率高、靈敏度高、準分子離子峰豐度高、碎片離子少等優點,還可以抗基質干擾,實現樣品的原位、無損、低碳、實時、直接和快速分析。使用DART離子源不需要繁雜冗長的樣品制備過程,極大地縮短了分析時間,節省了人力、物力資源,以實現實驗室高通量分析。近年來,利用DART-MS方法可對乳制品進行高通量和全自動的檢測,還經常用于火鍋底料和一些調味料中罌粟殼生物堿的檢測,操作過程簡單省時,為食品安全的監測工作帶來了有力的技術支持[20]。目前關于DART離子源質譜檢測喹諾酮類抗生素的研究很少。該研究通過對豬肉基質的溶劑提取和簡單凈化,DART離子源直接離子化,采用三重四極桿液質儀分析測定,實現喹諾酮類藥物殘留實時快速直接分析,建立一套獸藥殘留的快速篩查分析方法,以期為風險監測和監督抽檢工作提供指導方向。
1 材料與方法
1.1 試材與試劑豬肉(市售)。乙腈(色譜純,德國 Merck 公司); 5982-0032萃取鹽包 (美國Agilent公司); 5982-4950 凈化包(美國Agilent公司); PRiME HLB固相萃取小柱(美國waters公司); 有機微孔濾膜(孔徑0.22 μm,美國Agilent公司)。標準品:環丙沙星、達諾沙星、雙氟沙星、恩諾沙星、洛美沙星、氧氟沙星、奧比沙星、培氟沙星、沙拉沙星、司帕沙星(100 μg/mL,天津阿爾塔科技有限公司)。
1.2 儀器與設備電子天平(感量0.10 mg,METTLER TOLEDO ME104E;感量0.01 mg,METTLER TOLEDOXA205);Milli-Q去離子水發生器(美國Millipore公司);QL 866漩渦混合器(海門市其林貝爾儀器制造公司); SB-25-12DT超聲波發生器(寧波新芝生物科技有限公司);Velocity 18R Pro 離心機(澳大利亞 Dynamica 公司);N-Evap 氮吹儀(上海安譜實驗科技股份有限公司);AB 4500 三重四極桿液質儀 (美國AB SCIEX公司);DART SVP 離子源、樣品傳動軌道、進樣玻璃棒(美國Ion-Sense公司);隔膜泵(德國 Vacuubrand公司)。
1.3 試驗方法
1.3.1樣品前處理。市售豬肉均質勻漿后制成豬肉空白樣品。
直接提取法:稱取豬肉空白樣品2.5 g試樣于50 mL 塑料離心管中,加入10 mL乙腈,劇烈振蕩2 min。超聲提取20 min,于10 000 r/min離心5 min。取2 mL上清液,40 ℃下氮吹至干。加入0.5 mL 20%(體積分數)乙腈水復溶,渦旋30 s,超聲5 min充分溶解,過0.22 μm有機微孔濾膜,得到基質提取液,待測。
分散固相萃取凈化法:稱取豬肉空白樣品2.5 g試樣于50 mL塑料離心管中,加入10 mL乙腈,劇烈振蕩2 min。加入5 g萃取鹽包,渦旋,于10 000 r/min離心5 min。取6 mL上清液于5982-4950 凈化包中,渦旋30 s,4 000 r/min離心10 min。取2 mL上清液,40 ℃氮吹至干。加入0.5 mL 20%(體積分數)乙腈水復溶,渦旋30 s,超聲5 min充分溶解,過0.22 μm有機微孔濾膜,得到凈化包提取液,待測。
固相萃取柱凈化法:稱取豬肉空白樣品2.5 g試樣于50 mL塑料離心管中,加10 mL乙腈,劇烈振蕩2 min,于10 000 r/min離心5 min。取5 mL上清液轉移至PRiME HLB凈化小柱上,洗脫收集2 mL上清液,40 ℃氮吹至干。加入0.5 mL 20%(體積分數)乙腈水復溶,渦旋30 s,超聲5 min充分溶解,過0.22 μm有機微孔濾膜,得到凈化柱提取液,待測。
1.3.2儀器參數。質譜條件:掃描模式為全掃描模式(MRM);氣簾氣(CUR)137.9 kPa; 碰撞氣(CAD)為Medium;離子源溫度 (IHT)250 ℃;碰撞室脫離電壓 (CXP)6 V;碰撞室入口電壓(EP)10 V。DART 離子源條件:正離子模式;解離氣體溫度350 ℃;柵網電極電壓 200 V;樣品軌道傳動速度1.0 mm/s;隔膜泵壓力12.0 kPa。工作氣體和預備氣體均為高純度氮氣(99.999%),進樣裝置為玻璃棒進樣;用移液槍準確吸取液體2 μL于玻璃棒中,氦氣打在玻璃棒上,離子化之后進入質譜儀進行檢測。待測化合物定量離子對、定性離子對及碰撞能量見表1。

表1 待測化合物定量離子對、定性離子對及碰撞能量
1.3.3標準溶液配制。
1.3.3.1標準混標儲備液。分別量取1 mL環丙沙星、達諾沙星、雙氟沙星、恩諾沙星、洛美沙星、氧氟沙星、奧比沙星、培氟沙星、沙拉沙星、司帕沙星于10 mL容量瓶中,用乙腈定容并混勻,配制成10 μg/mL標準混標儲備液。
1.3.3.2標準混合中間液。取1 mL標準混標儲備液于10 mL 容量瓶中,用乙腈定容并混勻,配制成1 000 μg/L中間液。
1.3.3.3標準工作曲線。取10、20、50、80、100 μL標準混合中間液分別用乙腈、基質提取液、凈化包提取液、凈化柱提取液定容至1 mL,配制成10、20、50、80、100 μg/L標準濃度系列工作液。
1.3.4數據處理。取配制好的系列工作液進行測定,分別測定標準系列工作液定量離子相應的豐度,以標準工作液的濃度為橫坐標、豐度為縱坐標繪制標準曲線,外標法定量,數據采用Excel處理。
前處理過程總基質效應以ME值評價,基質效應計算參照文獻[21]的方法:
ME=B/A×100%
(1)
式中:ME為前處理過程總基質效應(%);A為目標化合物在純溶劑基質中檢測的峰面積;B為目標化合物在陰性樣品處理液基質中檢測的峰面積。
2 結果與分析
2.1 前處理凈化效果分析空白豬肉樣品按照“1.3.1”樣品前處理得到基質提取液、凈化包提取液、凈化柱提取液。取100 μL標準混合中間液分別用乙腈、基質提取液、凈化包提取液、凈化柱提取液定容至1 mL,按照“1.3.2”儀器參數對上述3種基質標進行測定,每個樣品測定4次,最終結果以平均值計,并按照公式(1)計算基質效應(ME),見表2。選取洛美沙星作為典型的提取離子流圖,見圖1。

圖1 0.1 μg/mL乙腈中洛美沙星提取離子流圖Fig.1 Ion flow diagram of lomefloxacin extraction in 0.1 μg/mL acetonitrile

表2 不同凈化方式的ME
基質效應(ME)可以反映基質效應的程度。通常ME>100%為基質增強效應,ME<100%為基質減弱效應,ME=100%則不產生基質效應。從表2可以看出,10 種化合物在豬肉中均受到不同的基質效應,在基質提取液中,其中環丙沙星、達諾沙星、培氟沙星、沙拉沙星這4種為增強效應,雙氟沙星、恩諾沙星、洛美沙星、氧氟沙星、奧比沙星、司帕沙星為抑制效應;環丙沙星受到基質增強效應最強,奧比沙星受到基質抑制效應最弱。在凈化柱提取液中,10 種化合物均為基質增強效應。在凈化包提取液中,除了奧比沙星外,其余均為基質增強效應。大多數化合物在凈化柱提取液和凈化包提取液中的ME均大于在基質提取液中的ME,這說明基質采取2種方法凈化均能夠提高ME,即凈化減少了雜質的含量,減少雜質與獸藥化合物競爭離子化,因而提高了獸藥化合物的離子化效率。此外,10種喹諾酮類化合物在凈化柱提取液ME大于凈化包提取液中的ME,這說明凈化柱的凈化效果優于凈化包,通過凈化柱能夠除掉更多的雜質,提高了獸藥化合物的離子化效率。
獸藥化合物在基質中的ME小于100%說明雜質與獸藥化合物競爭離子化,因而降低了獸藥化合物的離子化效率。獸藥化合物在基質中的ME大于100%,可能的原因是雜質與獸藥化合物離子化后,在離子源與質譜間的通道被吸附,雜質與獸藥化合物競爭吸附,從而抑制了獸藥化合物在通道的損失,增加了獸藥化合物通路,提高了響應值,當競爭吸附大于競爭離子化的作用,宏觀表現為ME大于100%。同一種基質經不同的方式凈化后,對喹諾酮類化合物產生不同的基質效應。這一方面可能是由于不同前處理方式的凈化效果和去除雜質種類不一致,導致目標化合物受到的基質干擾程度存在差異;另一方面可能是喹諾酮類化合物基本骨架均為氮雜雙并環,但不同化合物的分子和空間結構存在一定差異,可能導致其受到基質影響不同。
2.2 檢出限空白豬肉樣品按照“1.3.1”樣品前處理得到基質提取液、凈化包提取液、凈化柱提取液。取10 μL標準混合中間液分別用乙腈、基質提取液、凈化包提取液、凈化柱提取液定容至1 mL,按照“1.3.2”儀器參數對上述3種基質標進行測定,每個樣品測定4次,最終結果以平均值計,另取空白基質檢測,各化合物定量離子對響應結果見表3。
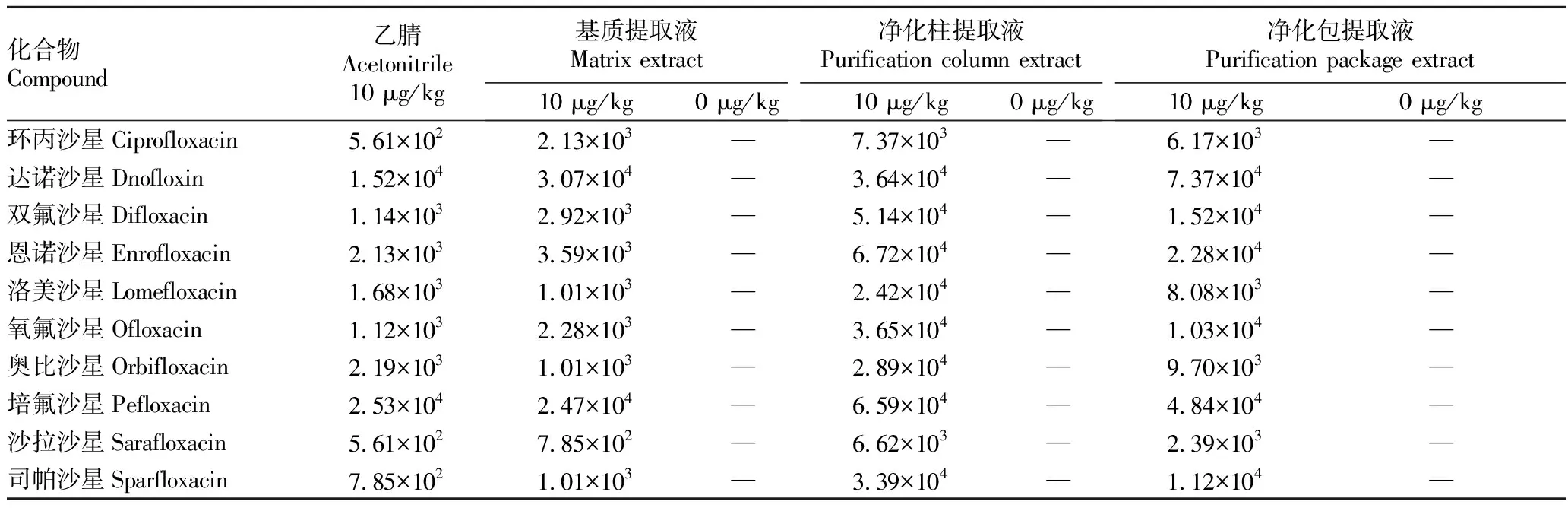
表3 檢出限及空白基質定量離子對響應
從表3可以看出,10種喹諾酮在10 μg/kg均有響應,在0 μg/kg時,基質提取液、凈化包提取液、凈化柱提取液中10種喹諾酮均沒有產生響應值。因此,直接使用基質提取液提取不進行凈化,10種喹諾酮均能夠在10 μg/kg檢出,滿足了GB 31650—2019 《食品安全國家標準 食品中獸藥最大殘留限量》判定要求,能夠實現不合格產品的快速篩查。
2.3 線性范圍取10、20、50、80、100 μL標準混合中間液分別用乙腈、基質提取液、凈化包提取液、凈化柱提取液定容至1 mL,配制10、20、50、80、100 μg/L標準濃度系列,按照“1.3.2”儀器參數對上述3種基質標進行測定,每個樣品測定4次,最終結果以平均值計,見表4。從表4可以看出,在10~100 μg/L,10種喹諾酮線性決定系數(R2)在0.700 0~0.970 0,線性關系良好。

表4 10種喹諾酮的線性方程、決定系數和線性范圍
2.4 精密度空白豬肉樣品按照“1.3.1”樣品前處理得到基質提取液、凈化包提取液、凈化柱提取液。取10、20、50、80、100 μL標準混合中間液分別用乙腈、基質提取液、凈化包提取液、凈化柱提取液定容至1 mL,配制10、20、50、80、100 μg/L 標準濃度系列,按照“1.3.2”儀器參數對上述3種基質標進行測定,每個樣品測定4次,最終結果以平均值計。精密度以相對標準偏差(RSD)計,10種喹諾酮的檢測精密度結果見表5。

表5 喹諾酮檢測精密度RSD
從表5可以看出,4種不同處理方式RSD為61.98%~185.26%, 溶劑提取液、基質提取液、凈化柱提取液、凈化包提取液的RSD平均值分別為112.36%、109.97%、69.70%、86.39%,凈化柱提取液RSD明顯低于其他基質的RSD,因此在檢測肉品中喹諾酮時,采用凈化柱凈化能夠獲得較好的精密度。
3 結論與討論
該研究通過對豬肉基質的溶劑提取和簡單凈化,并使用DART 離子源與三重四極桿質譜聯用技術(DART-QQQ),實現了10種喹諾酮實時快速直接分析。
該研究比較了基質提取液、凈化包提取液、凈化柱提取液中基質對目標物檢測的基質效應,結果表明凈化柱的凈化效果最好,能夠獲得較好的精密度。上述3種處理方法10種喹諾酮均能夠在10 μg/kg檢出,能夠實現不合格產品的快速篩查。10種喹諾酮在10 ~100 μg/L線性決定系數(R2)在0.700 0~0.970 0,線性關系良好。
因此,采用DART-QQQ檢測豬肉中10種喹諾酮作為快檢和對市售喹諾酮不合格豬肉的篩查,方法準確性、靈敏度好,并且能夠擴展更多種類獸藥高通量篩查,因此在多獸藥高通量快檢領域應用廣泛。
