基于模擬炮制研究滇烏堿砂炒轉化途徑及其前后毒效變化
2023-10-19 11:26:14劉叢改鄭清洋陳佳佳周園園王毓杰
中草藥 2023年20期
劉叢改,陶 培,鄭清洋,陳佳佳,周園園,陳 蓉,王毓杰
基于模擬炮制研究滇烏堿砂炒轉化途徑及其前后毒效變化
劉叢改,陶 培,鄭清洋,陳佳佳,周園園,陳 蓉,王毓杰*
成都中醫藥大學,四川成都 611137
研究滇烏堿在砂炒過程中的結構轉化途徑,比較滇烏堿與轉化產物的急性毒性和轉化產物的抗心律失常活性。采用HPLC法篩選出滇烏堿發生結構轉化的溫度和時間參數,通過柱色譜法和核磁共振(nuclear magnetic resonance,NMR)、質譜(mass spectrometry,MS)技術,分離鑒定出滇烏堿的砂炒轉化產物;采用急性毒性實驗比較原型成分和轉化產物的毒性強弱;采用烏頭堿建立大鼠心律失常模型,明確轉化產物的抗心律失常作用。通過比較不同炮制溫度、時間樣品的HPLC色譜圖,篩選出滇烏堿在140~180 ℃,加熱5~30 min時發生結構轉化;分離、鑒定出滇烏堿的砂炒產物pyroyunaconitine;急性毒性實驗結果表明,滇烏堿的半數致死量(lethal dose 50%,LD50)為0.353 mg/kg,pyroyunaconitine的LD50為62.563 mg/kg,炮制后毒性明顯降低。抗心律失常實驗結果顯示,隨著炮制產物pyroyunaconitine劑量的增大,大鼠的室性早搏(ventricular premature beat,VPB)潛伏期延長,室性心動過速(ventricular tachycardia,VT)發生率降低,心律失常完全抑制率逐漸升高,呈劑量相關性,表明其有一定的抗心律失常作用。砂炒過程中,滇烏堿的C-8位脫去乙酰基,生成去8-去乙酰滇烏堿;接著,8-去乙酰滇烏堿的C-8位和C-15位之間脫水形成雙鍵,生成pyroyunaconitine;pyroyunaconitine的毒性遠低于原型成分滇烏堿,并具有一定的抗心律失常活性。
烏頭屬;炮制;滇烏堿;砂炒;pyroyunaconitine;心律失常;急性毒性;HPLC;半數致死量
毛茛科(Ranunculaceae)烏頭屬L.植物在全世界有300余種,廣泛分布于北半球溫帶地區。我國分布有200余種,其中76種可供藥用[1-2];常用品種有烏頭Debx.、北烏頭Reichb.、短柄烏頭Diels.和甘青烏頭(Maxim) Stapf等。該屬植物中的主要成分為二萜生物堿,具有顯著的生理活性,已開發出氫溴酸高烏甲素片、乙酰烏頭堿片、草烏甲素片等鎮痛類藥物,用于緩解風濕痛、手術后疼痛等多種類型的疼痛[3]。
二萜生物堿也是烏頭類藥物的主要毒性成分,可引起嚴重的心律失常及呼吸中樞抑制,例如,烏頭堿可與心肌細胞、神經細胞膜上的鈉通道結合,使鈉通道持續開放,鈉離子內流增加,進而引起細胞膜電位失衡,導致心律失常、呼吸抑制等癥狀[4-5]。因此,為了確保用藥的安全性,烏頭類藥物需要經過炮制,才能用于臨床。常用的炮制方法包括:蒸法、煮法和砂炒法。前2種制法的炮制原理是:烏頭堿型有毒生物堿的C-8位、C-14位酯鍵在炮制過程中依次發生水解,生成毒性較低的單酯型生物堿和醇胺型生物堿,如烏頭堿轉化為苯甲酰烏頭原堿、烏頭原堿[6]。課題組研究證實,相對于蒸法和煮法,砂炒法能夠在更短的炮制時間內實現減毒目的,并且炮制原理與蒸法、煮法有明顯區別,毒性生物堿的結構轉化途徑更為豐富[7-10]。
滇烏堿(yunaconitine)是短柄烏頭、康定烏頭.Finet & Gagnep.中的主要二萜生物堿,其結構與印烏堿、草烏甲素相似,區別在于C-3位、C-4′位的取代基不同。滇烏堿的C-4′位為甲氧基,印烏堿的C-4′位為氫原子;滇烏堿的C-3位為羥基,草烏甲素的C-3位為氫原子。這些結構差異對滇烏堿在砂炒過程中的結構轉化過程及砂炒產物的毒性、活性會帶來哪些影響,尚不清楚。
本實驗以滇烏堿為例,以油浴的方式模擬砂炒炮制;采用色譜和波譜法對炮制產物進行分離鑒定,并通過藥材砂炒品進行驗證;采用急性毒性實驗比較滇烏堿及炮制產物的毒性大小;采用烏頭堿建立心律失常模型,明確炮制產物的抗心律失常活性,闡明砂炒過程中滇烏堿的結構轉化途徑及轉化產物的毒性和抗心律失常活性,為烏頭類藥物的炮制原理研究及抗心律失常的藥物研發提供思路。
1 儀器與材料
1.1 儀器
BL-420F型多功能生理記錄儀,成都泰盟軟件有限公司;島津LC-2010A型高效液相色譜儀,Lab Solutions色譜工作站,日本島津公司;ULUP-I-10T型優普超純水機,成都超純科技有限公司;CPA2250型電子分析天平,德國Sartorius公司;HH-SJ型集熱式磁力攪拌器,油浴用,金壇市城東新瑞儀器廠;MicrO-TOF-Q-II型質譜儀、Avance 600型核磁共振儀,瑞士Bruker公司;5型多功能炒貨機,常州市金壇邁斯機械有限公司。
1.2 藥品與試劑
滇烏堿(質量分數≥98.0%,本課題組從短柄烏頭中分離得到)、pyroyunaconitine(質量分數≥95.0%,本課題組從滇烏堿砂炒炮制品中分離得到),通過1H-NMR、13C-NMR和高分辨電噴霧電離質譜(high resolution electrospray ionization mass spectroscopy,HR-ESI-MS)確定結構;烏頭堿,批號WTZ10013,質量分數99.0%,陜西昊辰生物科技有限公司;8-去乙酰滇烏堿,批號DST230131-369,質量分數≥98.0%,成都德思特生物技術有限公司;鹽酸普羅帕酮(批號101190-201702,質量分數99.8%)、鹽酸利多卡因(批號100341-202104,質量分數93.1%),中國食品藥品檢定研究院;烏拉坦,批號2017090501,成都市科龍化工試劑廠;柱色譜硅膠,200~300目,青島海洋化工廠;生理鹽水,批號L122091801,四川科倫藥業股份有限公司;乙腈,色譜純,美國Fisher公司;水為超純水;其余試劑均為分析純;柱色譜所用石油醚沸點60~90 ℃;顯色劑為碘蒸氣。
短柄烏頭由西藏林芝市藏醫院提供,植物標本經成都中醫藥大學王毓杰副研究員鑒定,為短柄烏頭Diels.,憑證標本存放于成都中醫藥大學民族醫藥學院標本室。
1.3 動物
SPF級健康成年SD大鼠,雌雄兼用,7~8周齡,體質量(200±20)g;SPF級昆明小鼠,雌雄兼用,7~8周齡,體質量(18±2)g;均由北京斯貝福生物有限公司提供,生產許可證號:SCXK(京)2019-0010。實驗前進行適應性喂養2 d,自由飲水及進食,室溫維持在20~25 ℃,晝夜光照及通風環境自然調節,實驗前禁食12 h。動物實驗經成都中醫藥大學實驗動物倫理委員會批準,倫理備案號為2020-15。
2 方法與結果
2.1 滇烏堿的測定
2.1.1 色譜條件 色譜柱為Supersil ODS-B柱(250 mm×4.6 mm,5 μm);流動相為乙腈-0.03 mol/L碳酸氫銨溶液(濃氨水調pH 9.5),梯度洗脫:0~30 min,35%乙腈;30~70 min,35%~45%乙腈;70~90 min,45%乙腈;柱溫35 ℃;檢測波長260 nm;體積流量1.0 mL/min;進樣量10 μL。
2.1.2 對照品溶液的制備 取滇烏堿約100 mg,精密稱定,至250 mL量瓶,加二氯甲烷制成質量濃度為400.16 μg/mL的反應儲備液,取4 mL該反應儲備液,置10 mL量瓶中,揮干溶劑,加0.1%鹽酸甲醇溶液定容,制得質量濃度為160.06 μg/mL的對照品溶液,搖勻,過0.45 μm微孔濾膜,取續濾液,即得對照品溶液。
2.1.3 炮制品供試溶液的制備 取圓底燒瓶24個,分為120、140、160、180 ℃ 4個溫度組,每個溫度下設置6個炮制時間點(1、5、10、20、30、40 min)。分別精密加入“2.1.2”項下反應儲備液4 mL,揮干溶劑后,按照設置的溫度、時間進行炮制,反應結束后,放冷,加0.1%鹽酸甲醇溶液4 mL溶解,溶液轉移至10 mL量瓶中,圓底燒瓶再加0.1%鹽酸甲醇溶液洗2次,每次2 mL,洗液轉移至同一10 mL量瓶中,加0.1%鹽酸甲醇溶液定容至刻度,搖勻,過0.45 μm微孔濾膜,取續濾液,即得炮制品供試溶液。
2.1.4 線性關系考察 將準確稱量的滇烏堿溶解在0.1%鹽酸甲醇溶液中,制備線性儲備液,質量濃度達到2 mg/mL。在每個10 mL量瓶中分別精密加入儲備液0.025、0.100、0.200、0.400、0.800、1.000、2.000 mL,加0.1%鹽酸甲醇溶液定容至刻度,以制備最終質量濃度為5、20、40、80、160、200、400 μg/mL的對照品溶液。將上述對照品溶液進樣10 μL,按照“2.1.1”項下色譜條件,測定峰面積,并以進樣質量濃度為橫坐標(),峰面積為縱坐標(),構建標準曲線,進行線性回歸,得線性回歸方程為=119 56+184 57,=0.999 7,結果表明,滇烏堿在5~400 μg/mL線性關系良好。
根據3倍和10倍信噪比(/)所對應的滇烏堿質量濃度,確定該方法的檢測限為0.26 μg/mL,定量限為0.75 μg/mL。
2.1.5 精密度試驗 精密吸取同一質量濃度為160.06 μg/mL的滇烏堿對照品溶液,進樣量10 μL,連續進樣6次,測定滇烏堿的峰面積,計算其RSD值為0.45%,表明儀器精密度良好。
2.1.6 重復性試驗 取圓底燒瓶5個,分別加入“2.1.2”項下反應儲備液4 mL,揮干溶劑后,于160 ℃下炮制10 min,反應結束后,放冷,按“2.1.3”項下方法制備供試品溶液。按“2.1.1”項下色譜條件分別進行測定,進樣量10 μL,測定滇烏堿的峰面積,計算其質量分數的RSD值為2.59%,表明該方法的重復性良好。
2.1.7 穩定性試驗 精密吸取同一供試品溶液(160 ℃下炮制10 min)于0、2、4、6、8、10、12 h,按“2.1.1”項下色譜條件進行測定,進樣量10 μL,測定滇烏堿的峰面積,計算其RSD值為2.35%,結果表明供試品溶液在12 h內穩定性良好。
2.1.8 滇烏堿炮制前后含量測定 分別吸取“2.1.2”項下滇烏堿對照品溶液和“2.1.3”項下不同炮制溫度、不同炮制時間的供試品溶液10 μL,注入高效液相色譜儀,按照“2.1.1”項下色譜條件進行含量測定。從表1中可以看出炮制溫度越高,時間越久,滇烏堿的含量變化越明顯。例如,在160 ℃炮制5 min后含量降為106.79 μg/mL,160 ℃炮制30 min后含量降為14.79 μg/mL;180 ℃炮制5 min后含量降為7.78 μg/mL。
2.2 滇烏堿炮制轉化參數篩選
分別吸取滇烏堿對照品溶液及不同炮制品供試溶液10 μL注入液相色譜儀,得到不同溫度、不同時間條件下的色譜圖,能夠直觀反映出滇烏堿及其炮制產物的含量變化過程,結果見圖1、2。可知,炮制前的滇烏堿含量為160.06 μg/mL,在不同溫度下,隨著炮制時間的延長,滇烏堿的含量逐漸降低,并有新的色譜峰出現。溫度越高,時間越久,滇烏堿及炮制產物的含量變化越明顯。同時,從圖1可以看出,滇烏堿在140~180 ℃,炮制5~40 min后,在保留時間24、38、72 min處有較明顯的新色譜峰出現,提示在上述炮制溫度和炮制時間條件下,滇烏堿有3個炮制產物。在160 ℃炮制30 min時,炮制產物的數量較多,含量較高,因此,本實驗選擇在該溫度、時間條件下對滇烏堿的砂炒轉化產物進行分離制備。

表1 不同炮制品中滇烏堿含量

圖1 滇烏堿在不同炮制溫度和時間條件下的色譜圖

圖2 滇烏堿在不同溫度、時間條件下含量變化趨勢
2.3 滇烏堿砂炒轉化產物的分離及結構鑒定
2.3.1 柱色譜樣品的制備 取滇烏堿1.49 g,置250 mL圓底燒瓶中,加適量二氯甲烷溶解,用旋轉薄膜蒸發儀蒸干溶劑,使滇烏堿均勻地附著在圓底燒瓶內壁,將圓底燒瓶置于160 ℃下油浴30 min,取出,放涼,反應物加二氯甲烷溶解后,轉移至蒸發皿中,揮干溶劑,得到滇烏堿炮制產物(1.35 g),供上柱用。
2.3.2 分離純化 將滇烏堿炮制產物進行硅膠柱色譜(200~300目,130 g),分別用石油醚-丙酮-三乙胺10∶1∶0.01(2.2 L)、8∶1∶0.01(2.7 L)、6∶1∶0.01(4.2 L)、4∶1∶0.01(3 L)、2∶1∶0.01(2.1 L)、1∶1∶0.01(3 L)梯度洗脫,薄層板檢視,合并相近流分后,得到A~C 3個組分。組分A(0.5 g)進一步采用硅膠柱色譜(200~300目,130 g)分離純化,洗脫劑為石油醚-丙酮-三乙胺(6∶1∶0.01),得到砂炒產物1(200 mg)。
2.3.3 滇烏堿砂炒產物的結構鑒定 砂炒產物1:白色粉末,C33H45NO9。HR-ESI-MS/: 600.316 7 [M+H]+(計算值600.316 7),從NMR譜(表2)中可以看出,砂炒產物1屬于烏頭堿型二萜生物堿[11],具有1個--CH2-CH3(H1.07, 3H, t,= 7.2 Hz;H2.56, 2.64, 各1H, m;C13.5 q, 49.9 t),4個甲氧基 (H3.22, 3.27, 3.37, 3.29, 各3H, s;C56.4 q, 58.1 q, 57.2 q, 59.2 q),以及1個對甲氧苯甲酰基 (H7.98, 2H, d,= 8.4 Hz; 3.82, 3H, s; 6.88, 2H, d,= 8.4 Hz;C167.8 s, 122.8 s, 132.0 d (2C), 113.5 d (2C), 163.3 s, 55.4 q);H4.90 (1H, d,= 3.0 Hz) 歸屬于H-14β,表明對甲氧苯甲酰基連接于C-14位[12-13];HMBC遠程相關譜表明:1-OCH3(H3.22, s), 6-OCH3(H3.27, s), 16-OCH3(H3.37, s) 18-OCH3(H3.29, s) 分別與C-1 (C83.1, d), C-6 (C83.6, d), C-16 (C83.5, d), C-18 (C76.6, t) 相連(圖3);從13C-NMR譜中可以看出砂炒產物1具有9個含氧碳信號,因此,除了對甲氧苯甲酰基和4個甲氧基外,該化合物還具2個羥基,HMBC譜也表明C-3 (C71.9, d) 與H-2 (H2.17, 2.19), H-18 (H3.67, 3.77)相關,C-13 (C77.6, s) 與H-9 (H3.03), H-12β (H1.95), H-15 (H5.54) 相關。因此,這2個羥基分別位于C-3位和C-13位(表2)。

表2 砂炒產物1的1H-NMR和13C-NMR數據(600/150 MHz, CDCl3)

圖3 砂炒產物1的1H-1H COSY (?), HMBC (H→C) 和NOESY (?) 相關譜
砂炒產物1與滇烏堿的NMR圖譜相似,區別在于:(1)滇烏堿的C-8位有1個乙酰氧基,而該化合物在C-8位沒有乙酰氧基;(2)13C-NMR中,滇烏堿的C-8、C-15的化學位移分別為85.5和39.6[14],而砂炒產物1的C-8、C-15的化學位移分別向低場遷移到146.5和116.3。因此,1H-NMR中,H5.54 (1H, d,= 6.0 Hz) 歸屬為H-15,表明C-8和C-15之間形成了雙鍵[15-17]。根據1H-NMR、13C-NMR及2D-NMR(HMBC、1H-1H COSY、NOESY)數據(表2),可以確定砂炒產物1的結構為-乙基-8,15-雙去氫-14α-[(4′-甲氧基)苯甲酰氧基]-1α,6α,16β,18-四甲氧基-烏頭烷-3α,13β-二醇,即pyroyunaconitine。
2.4 短柄烏頭藥材砂炒品驗證實驗
2.4.1 色譜條件 色譜柱為Supersil ODS-B柱(250 mm×4.6 mm,5 μm);流動相為乙腈-0.03 mol/L碳酸氫銨溶液(濃氨水調pH 9.5),梯度洗脫:0~40 min,25%乙腈;40~80 min,25%~40%乙腈;80~100 min,40%~60%乙腈;100~120 min,60%~80%乙腈;柱溫35 ℃;體積流量1.0 mL/min;檢測波長260 nm。
2.4.2 對照品溶液的制備 取砂炒產物1約20 mg,精密稱定,至100 mL量瓶中,加0.1%鹽酸甲醇溶液溶解,定容,制得質量濃度為207 μg/mL的對照品溶液,搖勻,過0.45 μm微孔濾膜,取續濾液,即得砂炒產物1對照品溶液。
取8-去乙酰滇烏堿約5 mg,精密稱定,至10 mL量瓶中,加0.1%鹽酸甲醇溶液溶解,定容,制得質量濃度為520 μg/mL的對照品溶液,搖勻,過0.45 μm微孔濾膜,取續濾液,即得8-去乙酰滇烏堿對照品溶液。
2.4.3 砂炒炮制品的制備 將短柄烏頭藥材除去雜質、低溫烘干后切制成長度為1~2 cm的段,以適量干凈的河沙(粒度為10~24目,河沙用量以能夠完全淹沒藥材為度),在滾筒式炒藥機中加熱至160 ℃后,投入切制好的藥材,炒制10 min,轉速為15 r/min。
2.4.4 供試品溶液的制備 取短柄烏頭生品和砂炒品粉末(過7號篩)各2 g,精密稱定,加氨試液2 mL和乙醚60 mL,混勻,放置過夜,濾過,藥渣加乙醚40 mL,振搖1 h,濾過,藥渣再加乙醚洗3次,每次20 mL,濾過,合并濾液和洗液,低溫蒸干,殘渣加三氯甲烷2 mL溶解,轉入分液漏斗,用三氯甲烷3 mL洗容器,洗液并入分液漏斗中,用0.05 mol/L H2SO4提取3次,每次5 mL,合并酸液,酸液用三氯甲烷10 mL洗滌后,加氨試液調pH 至9左右,再用三氯甲烷提3次,每次10 mL,合并三氯甲烷液,三氯甲烷液用水20 mL振搖洗滌后,低溫蒸干,殘渣加0.1%鹽酸甲醇溶液溶解,定容至5 mL量瓶中,作為短柄烏頭藥材生品和砂炒品供試溶液,搖勻,過0.45 μm微孔濾膜,取續濾液,即得供試品溶液[18]。
2.4.5 短柄烏頭生品和砂炒品中滇烏堿及炮制產物的測定 分別吸取“2.4.2”項下滇烏堿、砂炒產物1和8-去乙酰滇烏堿對照品溶液10 μL和“2.4.4”項下短柄烏頭生品和砂炒品供試溶液10 μL,注入高效液相色譜儀,按照“2.4.1”項下色譜條件進行檢測。
2.4.6 短柄烏頭藥材砂炒品驗證結果 滇烏堿在生品中的質量分數為349.12 μg/g,在炮制品中下降至5.00 μg/g。8-去乙酰滇烏堿在生品中的質量分數為125.01 μg/g,在炮制品中上升至196.49 μg/g,炮制品中還出現了pyroyunaconitine的色譜峰,質量分數為38.60 μg/g。結果表明在砂炒過程中,滇烏堿依次轉化為8-去乙酰滇烏堿和pyroyunaconitine,結果見圖4。

A-滇烏堿 B-8-去乙酰滇烏堿 C-pyroyunaconitine D-短柄烏頭砂炒品 E-短柄烏頭生品
2.5 滇烏堿及其砂炒產物的毒性比較
2.5.1 實驗藥物的配制
(1)空白溶劑的配制方法:取1%鹽酸乙醇溶液4 mL,加生理鹽水稀釋,用5% NaOH調pH值至7左右,加生理鹽水定容至100 mL。
“離咱白石小學不遠,就是洪洞紅軍八路軍紀念館,抗日戰爭時期,朱德總司令曾經到白石村開展工作,與隨營學校的師生和白石村小學的學生打籃球,這些至今都是我們白石村的美談。我們白石小學至今還保存著朱總司令當時贈送的風琴、籃球架。”白石中心校程毅主任自豪地說。
(2)滇烏堿及砂炒產物1:取滇烏堿及砂炒產物1適量,加適量1%鹽酸乙醇溶液溶解,生理鹽水稀釋,用5% NaOH調pH值至7左右,加生理鹽水配制成不同質量濃度,充分混勻,備用。
2.5.2 急性毒性實驗 按預實驗結果選用SPF級昆明小鼠300只,雌雄各半,按體質量分層后隨機分為30組。每個受試化合物分別設置5~8個劑量組(組間距=0.8~0.9),同時設置溶劑對照組。每只小鼠均按照0.015 mL/g尾靜脈給藥1次,分別觀察給藥后14 d內出現的中毒癥狀,死亡情況及死亡時間,對觀察期滿或中毒死亡的小鼠進行解剖,肉眼觀察主要臟器的改變。
按Bliss方法計算動物的半數致死量(lethal dose 50%,LD50)及其95%的可信限。
2.5.3 滇烏堿及砂炒轉化產物的毒性比較結果 課題組前期已完成印烏堿及其轉化產物的心臟毒性研究,為了明確滇烏堿的C-4′甲氧基對急性毒性帶來的影響,除了滇烏堿和pyroyunaconitine之外,本實驗增加了印烏堿及其砂炒產物mithaconitine和Δ15(16)-16-demethoxyindaconitine的急性毒性研究。不同受試藥物組小鼠在給藥后大多數于1~10 min內死亡,所有動物均出現呃逆、舔足、流涎等癥狀,嚴重者可見跳躍、角弓反張、小便失禁。
Pyroyunaconitine組有部分小鼠出現口鼻、四肢及尾巴變紅、便血、瞳孔變白的癥狀,少數動物解剖后出現胃部脹氣、胸腔及肺部瘀血,其余器官無肉眼可見異常。不同受試化合物的LD50及95%可信限見表3。
2.6 砂炒產物1的抗心律失常活性研究
取SD大鼠98只,隨機分為隨機分為空白溶劑組、模型對照組、陽性對照組(普羅帕酮、利多卡因)、砂炒產物1不同劑量組(0.4、0.6、0.8 mg/kg)7組。空白溶劑的配制方法:取1%鹽酸乙醇溶液4 mL,加生理鹽水稀釋,用5% NaOH調pH值至7左右,加生理鹽水定容至100 mL。烏頭堿、陽性藥和受試藥物均采用空白溶劑的配制方法進行配制。
SD大鼠ip 20%烏拉坦(劑量1.2 g/kg[19])麻醉,仰位固定,針型電極插入四肢皮下,使用BL-420F多功能生理記錄儀觀察大鼠正常II導聯心電圖20 min后,暴露股靜脈,從股靜脈分別注射相應劑量的受試藥物、陽性藥和等體積空白溶劑,10 min后再從股iv烏頭堿(0.03 mg/kg)建立心律失常模型[20-21],記錄30 min內各組大鼠第1次出現室性早搏(ventricular premature beat,VPB)的時間(VPB潛伏期)、是否出現心動過速以及心律失常的發生率。
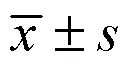
2.6.1 Pyroyunaconitine對烏頭堿誘發大鼠VPB潛伏期的影響 空白溶劑組大鼠在整個觀察期內,心電圖未見異常。模型組大鼠經股iv烏頭堿后出現典型的室性早搏(ventricular premature beat,VPB)、室性心動過速(ventricular tachycardia,VT),并有一部分動物出現室顫(ventricular fibrillation,VF),持續30 min以上,表明心律失常模型建立成功。模型組的VPB潛伏期為(105.6±28.3)s,普羅帕酮組為(201.7±77.0)s,利多卡因組為(208.5±69.6)s;與模型組相比,2種陽性藥均有顯著性差異(<0.05)。Pyroyunaconitine不同劑量組(0.4、0.6、0.8 mg/kg)的VPB潛伏期分別為(187.1±93.7)、(229.5±77.8)、(419.1±166.1)s;與模型組相比,0.6 mg/kg和0.8 mg/kg劑量組均有顯著差異(<0.05);與普羅帕酮及利多卡因組相比,0.8 mg/kg劑量組有顯著差異(<0.05),結果如表4所示。

表3 不同受試化合物的LD50
2.6.2 Pyroyunaconitine對烏頭堿誘發大鼠VT發生率的影響 如果VPB沒有得到及時治療,將會進一步發展為VT,VT的心電圖特征為連續出現3次或3次以上的VPB,QRS波群寬大畸形,無恒定的P波[24]。VT發生率可用于評價藥物的抗心律失常作用,VT發生率越低,抗心律失常作用越好。
2檢驗結果顯示,3個劑量組之間的VT發生率無顯著性差異(>0.05),但從表5中可以看出,隨著劑量的增加,VT發生率逐漸降低(由0.4 mg/kg組的100%降為0.8 mg/kg組的56.5%)。

表4 Pyroyunaconitine對烏頭堿誘發大鼠VPB潛伏期的影響()
與模型組比較:*<0.05;與普羅帕酮組比較:#<0.05;與利多卡因組比較:Δ<0.05
P< 0.05model group;P< 0.05propafenone group;Δ< 0.05lidocaine group
2.6.3 Pyroyunaconitine對大鼠心律失常完全抑制率的影響 心律失常發生率[25-26]是指大鼠預先注射受試藥物,再用烏頭堿建立心律失常模型后,30 min內出現心律失常(包括VPB、VT、VF)的比例。心律失常完全抑制率=1-心律失常發生率,即30 min內未出現心律失常的比例,可以評價受試藥物能否完全拮抗烏頭堿的致心律失常作用。
根據2檢驗的要求,將受試藥物的0.4 mg/kg和0.6 mg/kg 2個劑量組合并為0.4~0.6 mg/kg劑量組。總的卡方檢驗結果顯示,0.4~0.6 mg/kg組、0.8 mg/kg組及利多卡因組的心律失常完全抑制率有差異(<0.05),結果如表6所示。兩兩比較結果顯示,0.4~0.6 mg/kg劑量組的心律失常完全抑制率為4.2%,顯著低于利多卡因組(<0.05);0.8 mg/kg劑量組的心律失常完全抑制率為17.4%,與利多卡因組沒有顯著性差異(>0.05)。0.4~0.6 mg/kg劑量組與0.8 mg/kg劑量組的心律失常完全抑制率雖然沒有顯著性差異(>0.05),但隨著劑量的增加,抑制率逐漸增大,呈現一定的量效關系。結果如表7所示。
3 討論
通過HPLC法比較不同炮制溫度、炮制時間條件下滇烏堿的結構轉化情況,發現在160 ℃下炮制30 min時炮制產物的數量較多,含量較高。因此,選擇該溫度、時間條件制備滇烏堿炮制品,并采用柱色譜和波譜法,分離鑒定出炮制產物pyroyunaconitine。

表5 Pyroyunaconitine的VT發生率χ2分析
表6 Pyroyunaconitine心律失常完全抑制率χ2分析

表7 Pyroyunaconitine對烏頭堿誘發大鼠心律失常完全抑制率的影響
與利多卡因組比較:*<0.05
P< 0.05lidocaine group
驗證實驗證實藥材砂炒品中8-去乙酰滇烏堿的含量較生品增加,并且出現了pyroyunaconitine。與滇烏堿相比,pyroyunaconitine的C-8位沒有乙酰基,C-8位和C-15位之間形成了雙鍵;通過結構對比,可以推測出滇烏堿在炮制過程中的轉化途徑之一為:滇烏堿的C-8位酯鍵裂解,生成8-去乙酰滇烏堿;8-去乙酰滇烏堿的C-8位羥基和C-15位氫原子,發生脫水反應形成雙鍵,生成pyroyunaconitine。反應過程如圖5所示。
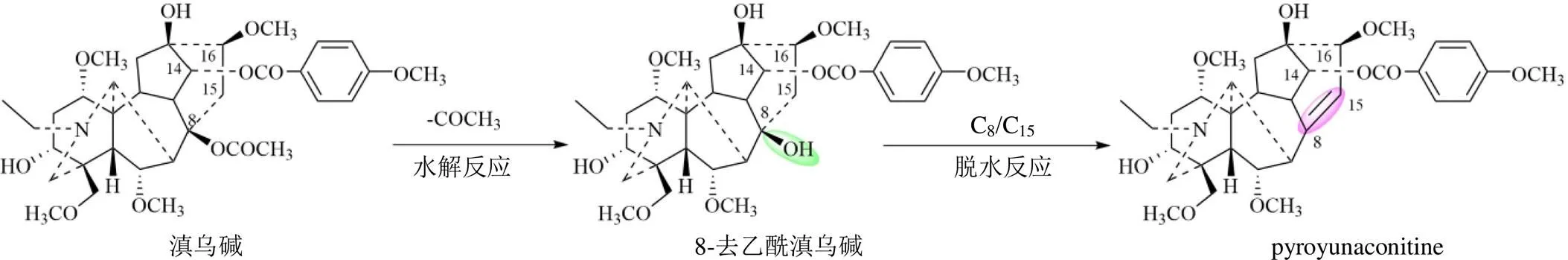
圖5 滇烏堿在砂炒過程中的轉化途徑
急性毒性實驗結果顯示滇烏堿的LD50為0.353 mg/kg,pyroyunaconitine的LD50為62.563 mg/kg,是滇烏堿的177倍;類似的,印烏堿的LD50為0.453 mg/kg,炮制產物mithaconitine和Δ15(16)-16- demethoxyindaconitine的LD50分別為42.323 mg/kg和9.013 mg/kg,是印烏堿的93倍和20倍。上述結果表明,炮制產物的急性毒性均遠低于原型成分,不同結構的炮制產物相比,具有C-8和C-15位雙鍵的炮制產物毒性更低,這與草烏甲素炮制產物pyrocrassicauline A的結果一致[9]。
滇烏堿和印烏堿的結構差異在于C-4′位取代基不同,從LD50值可以看出,滇烏堿的急性毒性大于印烏堿,表明4′-OCH3能夠增加該類成分的毒性。滇烏堿和草烏甲素的結構差異在于C-3位取代基不同,草烏甲素的LD50為0.659 mg/kg[9],大于滇烏堿的LD50值,表明該類型化合物的3-OH能夠增加毒性。再結合印烏堿的LD50值可以進一步推測出,3-OH增加急性毒性的作用大于4′-OCH3。
抗心律失常活性實驗采用烏頭堿建立大鼠心律失常模型,其機制為烏頭堿激活心肌細胞膜的鈉通道,使其處于開放狀態,鈉離子內流增加,動作電位時程延長,引起心律失常。鈉通道阻斷劑分為Ia、Ib和Ic類。其中,Ib類的利多卡因和Ic類的普羅帕酮是臨床常用的抗心律失常藥物,可以阻斷烏頭堿的作用,廣泛用于治療室性早搏、室性心動過速等心律失常。因此,本實驗選擇了利多卡因和普羅帕酮作為陽性藥。
以VPB潛伏期、VT發生率、心律失常完全抑制率作為評價受試藥物抗心律失常活性的指標,VPB潛伏期和VT發生率均證實pyroyunaconitine具有較好的抗心律失常作用。與VPB潛伏期和VT發生率相比,心律失常完全抑制率更能直接反映出受試藥物的抗心律失常作用強弱,抑制率越高,說明受試藥物的作用越好。陽性藥利多卡因組的抑制率為37.5%,與0.8 mg/kg組的17.4%沒有顯著性差異(>0.05),但顯著高于0.40~0.60 mg/kg組的4.2%(<0.05)。綜合考慮上述指標的結果,可以看出pyroyunaconitine的抗心律失常作用雖然弱于利多卡因,但是在延長VPB潛伏期方面具有一定的優勢。本實驗通過比較滇烏堿及pyroyunaconitine的結構,梳理出滇烏堿在砂炒過程中的結構轉化途徑。通過整體動物實驗證實,pyroyunaconitine的毒性遠低于滇烏堿,并有一定的抗心律失常作用。研究結果對于烏頭類藥物砂炒炮制原理的闡明、砂炒工藝的規范和質量標準的完善具有積極作用。
利益沖突 所有作者均聲明不存在利益沖突
[1] 中國科學院中國植物志編輯委員會. 中國植物志 (第二十七卷) [M]. 北京: 科學出版社, 1979: 113.
[2] 肖培根, 王鋒鵬, 高峰, 等. 中國烏頭屬植物藥用親緣學研究 [J]. 植物分類學報, 2006, 44(1): 1-46.
[3] 高黎明, 毛學峰, 魏小梅, 等. 二萜類生物堿的藥理作用及構效關系研究概況 [J]. 西北師范大學學報: 自然科學版, 1999, 35(1): 98-103.
[4] Wang F P, Chen Q H, Liu X Y. Diterpenoid alkaloids [J]., 2010, 27(4): 529-570.
[5] Ameri A, Shi Q, Aschoff J,. Electrophysiological effects of aconitine in rat hippocampal slices [J]., 1996, 35(1): 13-22.
[6] 鐘凌云. 中藥炮制學 [M]. 第5版. 北京: 中國中醫藥出版社, 2021: 322-324.
[7] Wang Y J, Tao P, Wang Y. Attenuated structural transformation of aconitine during sand frying process and antiarrhythmic effect of its converted products [J]., 2021, 2021: 7243052.
[8] Wang Y J, Wang Y, Tao P. Structural characterization,toxicity and biological activity of two new pyro-type diterpenoid alkaloids derived from 3-acetylaconitine [J]., 2023, 21(3): 302-314.
[9] Tao P, Wang Y, Wang Y J. Attenuation and structural transformation of crassicauline A during sand frying process and antiarrhythmic effects of its transformed products [J]., 2021, 12: 734671.
[10] Wang Y, Tao P, Wang Y J. Attenuated structural transformation of indaconitine during sand frying process and anti-arrhythmic effects of its transformed products [J]., 2022, 2022: 8606459.
[11] Li Y Z, Qin L L, Gao F,. Kusnezosines A-C, three C19-diterpenoid alkaloids with a new skeleton fromReichb. var.[J]., 2020, 144: 104609.
[12] Pelletier S W, Joshi B S. Carbon-13 and proton NMR shift assignments and physical constants of norditerpenoid alkaloids [A] // Pelletier S W.:[M]. New York: Springer, 1991: 297-564.
[13] Gao F, Chen D L, Wang F P. Two new C19-diterpenoidalkaloids fromvar.[J]., 2006, 54(1): 117-118.
[14] 王建莉, 周先禮, 王鋒鵬. 秦嶺翠雀花中生物堿成分的研究 [J]. 天然產物研究與開發, 2003, 15(6): 498-501.
[15] Desai H K, Hart B P, Caldwell R W,. Certain norditerpenoid alkaloids and their cardiovascular action [J]., 1998, 61(6): 743-748.
[16] Yue J M, Xu J, Chen Y Z,. Diterpenoid alkaloids from[J]., 1994, 37(5): 1467-1470.
[17] Yue J M, Chen Y Z, Li Y Z. C19-diterpenoid alkaloids of[J]., 1990, 29(7): 2379-2380.
[18] 王毓杰, 曾陳娟, 姚喆, 等. 民族藥鐵棒錘不同藥用部位中生物堿含量測定 [J]. 中成藥, 2010, 32(8): 1390- 1393.
[19] Bai D L, Chen W Z, Bo Y X,. Discovery of-(3, 5-bis(1-pyrrolidylmethyl)-4-hydroxybenzyl)-4-methoxybenzenesulfamide (sulcardine) as a novel anti-arrhythmic agent [J]., 2012, 33(9): 1176-1186.
[20] 王艷, 陶培, 王毓杰, 等. 油浴模擬砂炒過程中印烏堿的結構轉化途徑及轉化產物毒性研究 [J]. 中草藥, 2020, 51(5): 1205-1213.
[21] 徐叔云. 藥理實驗方法學[M]. 第3版. 北京: 人民衛生出版社, 2002: 1173-1174.
[22] Hong B H, He J L, Le Q Q,. Combination formulation of tetrodotoxin and lidocaine as a potential therapy for severe arrhythmias [J]., 2019, 17(12): 685.
[23] 李紅, 牛欣, 李國彰, 等. 調脈飲注射液抗心律失常的實驗研究 [J]. 中國中藥雜志, 2006, 31(9): 759-762.
[24] Brady W J, Mattu A, Tabas J,. The differential diagnosis of wide QRS complex tachycardia [J]., 2017, 35(10): 1525-1529.
[25] Bartosova L, Novak F, Bebarova M,. Antiarrhythmic effect of newly synthesized compound 44Bu on model of aconitine-induced arrhythmia: Compared to lidocaine [J]., 2007, 575(1/2/3): 127-133.
[26] 王培德, 馬學民, 張慧靈, 等. 刺烏頭堿對麻醉大鼠心電圖的影響及其抗實驗性心律失常作用 [J]. 中國藥理學通報, 1997, 13(3): 263-265.
Transformation pathway of yunaconitine and toxic-efficacy changes before and after stir-frying based on simulation processing technology
LIU Cong-gai, TAO Pei, ZHENG Qing-yang, CHEN Jia-jia, ZHOU Yuan-yuan, CHEN Rong, WANG Yu-jie
Chengdu University of Traditional Chinese Medicine, Chengdu 611137, China
To study the structural transformation pathway of yunaconitine during the stir-frying process with sand, and compare the acute toxicity and antiarrhythmic activity of yunaconitine with its processing products.Using HPLC to screen out the temperature and time parameters of the structural transformation of yunaconitine; The processing products of yunaconitine were separated and identified using column chromatography, nuclear magnetic resonance (NMR) and mass spectrometry (MS); The acute toxicity test was performed to compare the toxicity of yunaconitine with its processing products; The aconitine-induced arrhythmia model was used to clarify the activity of the processing products.Yunaconitine was converted into pyroyunaconitine when it was heated at 140—180 ℃ for 5—30 min. The acute toxicity test results showed that the lethal dose 50% (LD50) of yunaconitine and pyroyunaconitine were 0.353 mg/kg and 62.563 mg/kg, respectively. The toxicity of yunaconitine was significantly reduced after processing. In the antiarrhythmic assay, the pyroyunaconitine could delay the onset time of ventricular premature beat (VPB), reduce the incidence of ventricular tachycardia (VT) and increase arrhythmia inhibition rate with increasing dose. The result indicated that pyroyunaconitine dose-dependently exhibited antiarrhythmic activities.The structural transformation pathway of yunaconitine has been defined: The acetoxyl group at C-8 of yunaconitine was firstly hydrolyzed to a hydroxyl group, and then a double bond was subsequently introduced at C-8/C-15 via further dehydration, resulting in the formation of pyroyunaconitine. Compared with yunaconitine, the toxicity of pyroyunaconitine was reduced. And it possesses specific antiarrhythmic properties.
L.; processing; yunaconitine; stir-frying with sand; pyroyunaconitine; arrhythmia; acute toxicity; HPLC; lethal dose 50%
R283.6
A
0253 - 2670(2023)20 - 6671 - 11
10.7501/j.issn.0253-2670.2023.20.011
2023-05-19
四川省科技計劃項目(2020YJ0131);成都中醫藥大學“杏林學者”學科人才科研提升計劃(QJRC2022046)
劉叢改(1999—),女,碩士研究生,主要方向為中藥新制劑、新劑型、新技術。Tel: 15649367318 E-mail: 1160743059@qq.com
通信作者:王毓杰(1980—),男,副研究員,碩士生導師,研究方向為中藥及民族藥炮制原理。Tel: 18980923679 E-mail: superwangyj@126.com
[責任編輯 鄭禮勝]
