第35屆中國化學奧林匹克(初賽)試題解析
——以有機化學部分第9、10題為例
2023-10-07 10:37:10楊明岸李俊吳敏鍶張喜庭
大學化學 2023年8期
關鍵詞:化學
楊明岸,李俊,吳敏鍶,張喜庭
1 廣東第二師范學院化學系,廣州510303
2 華南師范大學化學學院,廣州 510006
3 廣州大學化學化工學院,廣州510006
第35屆中國化學奧林匹克(初賽)在全國28個省、自治區、直轄市舉行,約7萬名中學生參與了本次競賽[1]。作為全國性的化學賽事,競賽鼓勵青少年接觸化學發展的前沿,激發學生鉆研化學的興趣,探索化學家的思想淵源與工作方法,為國家選拔化學理論功底扎實、能與化學家在研究思路上產生“共振”的科學型人才創造了機遇與條件[2]。總體來看,試題的綜合難度與往屆持平。相較于第34屆試題,在知識板塊的分布上,分值比重明顯增加的是無機化學(含結構化學,如分子結構、晶體結構);明顯減小的是高分子化學、配位化學與分析化學;基本持平的是元素化合物、有機化學與物理化學。初賽的基本內容仍集中在無機化學(含元素化合物)和有機化學這兩個部分,分數約占全卷的60%[3]。有機化學模塊在試題的設置上穩中求進,側重于考查學生對基礎知識的理解與運用能力,部分內容選材于近兩年有機化學領域較新并承載重要意義的研究成果。此外,試題強化了空間力、推理力與信息分析能力這三個維度的要求。通過對第35屆化學初賽第9、10題的分析(見表1),幫助處于啟蒙階段的競賽選手熟悉有機化學模塊的試題結構與考查風格,為從事化學競賽培訓的教師提供參考。第35屆中國化學奧林匹克(初賽)有機化學部分第9、10題原題已放入補充材料,讀者可從參考文獻[1]或本文的補充材料快速獲得競賽題目。

表1 第35屆化學奧林匹克(初賽)第9、10題考情細目表
1 第9題解析
1.1 考題解析
[9-1] 本題考察吡啶類衍生物與酸酐、三級膦之間的反應。反應的轉化過程與第34屆初賽第10題的Pummerer重排較接近[2]。二芳基三氟甲基膦是一種親核試劑。結合產物結構看,它不與Tf2O (三氟甲磺酸酐)反應,而是對吡啶進行三氟甲基化。Tf2O起到活化吡啶環的作用,環上的N原子為親核位點,Tf2O的S原子為親電位點,發生加成反應生成中間體M1。在O-的參與下,通過給電子對的轉移,TfO-作為離去基團發生消除反應。二芳基三氟甲基膦親核進攻中間體M2的C(4)進行1,4-加成反應,生成具有醌式結構的季鏻鹽,為吡啶結構的三氟甲基化提供了可能性。中間體M3的C(4)的質子被大位阻堿DBU (1,8-二氮雜雙環[5.4.0]-7-十一碳烯)[4]攫取轉化為具有親核能力的反應位點,在給電子對的作用下,N-S鍵斷裂使Tf-基團離去。由于是強酸性水解的條件,中間體M4吡啶環上的N在質子化的同時,磷原子結合水分子中的羥基。經分子內重排,離去二芳基羥基膦,最后得到三氟甲基化產物。反應的歷程如圖1所示,A和B的結構簡式見圖2。

圖1 吡啶C4位三氟甲基化的反應機理

圖2 化合物A和B的結構簡式
[9-2] 本題考察Claisen重排反應與Alder-ene反應在成環中的應用。反應條件是330 °C的高溫,無外加試劑,可聯想到周環反應。底物是一個烯丙基乙烯基醚類衍生物結構,是典型的[3,3]-σ重排的骨架體系,故由反應物轉化到中間體M5發生的是Claisen重排反應(圖3標注的兩個M5是同一種中間體,即題目要求書寫的A。為表示清楚反應歷程,凸顯出不同位置的H原子)。反應經歷六元環過渡態的分子內重排,烯氧基在五元環紙面朝下,所以協同反應產生的乙醛基仍在五元環紙面朝下。在完成[3,3]-σ重排后,發生周環反應。周環反應過程涉及的位點如圖3所示(紅色突出)。從M5到pdt-1,存在的轉化有:① H原子由C(5)轉移到C(9);②π鍵斷裂:C(1)-C(2),C(8)-C(9);③ 新鍵形成:σ鍵C(2)-C(8),π鍵C(1)-C(5);從M5到pdt-2,存在的轉化有:① H原子由C(10)轉移到C(1);②π鍵斷裂:C(1)-C(2),C(8)-C(9);③ 新鍵形成:σ鍵C(2)-C(8),π鍵C(9)-C(10)。根據上述信息發現這與一般的周環反應類型如電環化、σ遷移的特征不符,與環加成(如D-A反應)具有相當的共性。本題中,電子從烯體(含烯丙基氫的富電子基團)的HOMO流向親烯體(含π鍵的缺電子基團)的LUMO。過程涉及H原子與烯體雙鍵的遷移,在親烯體不飽和鍵的兩端構建兩個新的σ鍵,同時雙鍵移動至原烯丙基位置,這一轉化路徑被稱為Alder-ene反應(烯反應),屬于六電子的周環過程。C(1)-C(2),C(8)-C(9)這兩個雙鍵的α-C上均有一個H原子,進行烯反應時兩個雙鍵都具備提供烯丙基氫的潛在性,故存在兩種產物:若C(2)-C(1)-C(5)體系作為烯體,C(8)-C(9)作為親烯體,生成物是pdt-1;若C(8)-C(9)-C(10)體系作為烯體,C(1)-C(2)作為親烯體,生成物是pdt-2。本題中烯反應的環狀過渡態為穩定的船式構象TS-1和TS-2[5],分別對應題目的過渡態B和C;M5對應題目的中間體A,轉化過程見圖3。

圖3 烯丙基乙烯基醚類衍生物在高溫條件下進行Claisen重排與Alder-ene反應的機理
[9-3] 本題涉及親核取代反應、核磁共振氫譜和芳香性等重要知識。一般來說,進攻的親核試劑的親核能力越強,反應經過SN2機理過渡態所需的活化能就越低,SN2反應趨向越大。AgNO3是一種弱的親核試劑,故傾向于進行SN1反應。反應物中的Cl原子被Ag+以靜電作用誘導離去,生成的碳正離子被-ONO親核進攻得到了產物硝酸酯[6,7]。而MeLi是一種強的親核試劑,傾向于進行SN2反應。
根據題意“分子式為C9H12的產物”,結合反應物的分子式C8H9Cl后可推斷出氯原子被換作甲基。“產物的1H-NMR譜表明,在化學位移0.89 ppm處有六個氫”可分析出:① 由于六個氫原子是化學等價的,產物中很可能存在兩個化學環境一致的甲基;② 質子吸收峰處于0.89 ppm的信號位置是光譜中屏蔽效應很強(可通俗理解成質子對外磁場的感受少)的高場區域。從數值上看,甲基所連基團的給電子能力應比較強。反應物中已有一個甲基,可推斷出第二個甲基是由試劑MeLi親核進攻得到的。甲基負離子(Me-)的加成將得到碳負離子中間體,可將甲基負離子加成到各種位置,對得到的各種碳負離子中間體進行比較。如圖4所示:由Hückel規則得知,環戊二烯負離子具有芳香性,穩定性好,證明了Me-進攻C(6)的反應傾向于發生。接著環戊二烯負離子進攻C(8)-Cl鍵使其異裂,Cl-離去,發生分子內關環。產物中兩個甲基均連在環丙基上,印證了上述的推斷②。
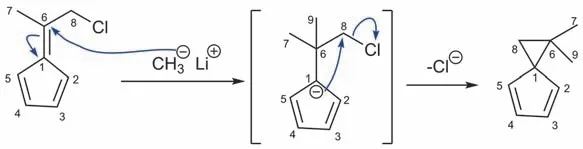
圖4 環戊二烯衍生物和甲基鋰發生SN2反應的機理
1.2 評注與拓展
[9-1] 本題可從分子式與價態的角度思考:將兩個底物的分子式相加并與A的分子式比較。A相比于底物多了1個C,1個S,3個F,3個O原子;少了1個H原子。多出的原子部分可恰好組成TfO-,作為A的陰離子。減少的氫質子很可能被DBU捕獲,形成DBUH+作為B的陽離子(DBUH+的分子式不符合A中陽離子的要求)。由“吡啶的C4位三氟甲基化”可知,二芳基三氟甲基膦傾向于親核進攻C(4),在吡啶環上引入鏻鎓離子。此時P(III)變為了P(V),發生氧化反應。相應地,Tf2O就被還原成Tf-,可作為B的陰離子。經檢驗,這一推斷滿足得失電子守恒。A的陽離子骨架不難推斷,可看成是鏻鎓離子取代吡啶C4位的H原子。
題目選材于2021年4月發表于Nature的一項研究成果[8]。科羅拉多州立大學的McNally課題組通過磷介導的sp2-sp3配體偶聯反應,無需預官能團化和導向基團,便可通過“一鍋法”直接將吡啶結構中的C-H鍵氟烷基化。該反應的選擇性高,在復雜分子的后期功能化上展現出誘人的應用前景。
[9-2] Claisen重排反應的機理[9]如圖5所示(該圖不表達任何特定的前線軌道,只體現構象中新的成鍵與相應軌道的聯系),這種[3,3]-σ遷移重排一般經過類椅式過渡態[10]910。

圖5 Claisen重排反應的機理
化學家Alder在1943年首次提出Alder-ene反應。他通過高溫加熱丙烯和馬來酸酐兩種底物,合成了烯丙基琥珀酸酐,該過程可視為丙烯(含烯丙基氫)和馬來酸酐發生了間接取代加成反應[11]。Alderene反應在大多數教科書中被歸為環加成反應,在有些教科書中被獨立劃分為第四類周環反應[10]894-895,第30屆決賽理論試題的[8-2]就涉及了這種反應[12]。看待Alder-ene反應最簡單的方式,是將它轉化為一類熟悉的環加成反應:D-A反應,其中雙烯的一個π鍵被一個C-H鍵替代,產物形成一個新的C-C鍵與一個跨空間轉移的H原子,反應機理見圖6[13,14]。與Diels-Alder反應不同,Alder-ene反應中C-Hσ鍵的斷裂要求更高的活化能,反應條件較為苛刻。但因其廣泛的底物適用性[15],是一種用于高效構建C-C或C-X鍵(X:雜原子)的策略,在一些天然產物與復雜分子的合成中承擔著重要的功能。

圖6 Alder-ene反應的機理
[9-3] 核磁共振(NMR)波譜是化學家們進行結構解析與研究相互作用最常用的重要工具,是少數能用于無損檢測三種物態的技術之一,靈敏度高(能分析質量不足1 mg樣品的結構)。在解析物質的結構時,通常需要借助多個核磁實驗從不同角度來展示分子中原子核及其核外電子的磁特性,據此信息推導出相應的分子結構。可以說,幾乎所有的有機或生物分子以及許多無機分子的結構解析都源于NMR波譜技術[16]。自問世半個多世紀以來,NMR歷經多次技術革新,這一領域的重大進展日新月異。本屆初賽試題沒有對NMR波譜單獨命題,而將其作為輔助信息融入題干,彰顯現代儀器定性分析的表征手段。“化學理論與實驗結合”這一思想的滲透,是試題的特色之處。
2 第10題解析A
2.1 考題解析
[10-1] 本題考察酰胺共振式的書寫。共振式之間的差別體現在電子的排布。書寫分子的共振式時要注意:① 共振式要符合Lewis結構式;② 代表同一分子的共振式具有相同的原子排列順序且具有相等的未成對電子數[17]。普通酰胺A的共振式可仿照烯醇互變異構書寫,如圖7采用共平面的畫法。

圖7 酰胺的共振式
[10-2-1] 本題考察端基效應與基團的電負性、離去能力之間的關系。基團Y的孤對電子偏移至N-Y之間,從而促使基團Z離去。Z基團的吸電子能力越強,越容易離去,使酰胺N原子的正電性增強,親核試劑更容易與其發生取代反應,端基效應更強。本題中,基團的吸電子能力可通過電負性衡量,電負性有:F > O > N,即基團的端基效應有:F-> OR-> NR-2。故三種雜原子中,F原子取代酰胺具有最強的端基效應。
[10-2-2] 本題考察不同類型電子效應的競爭作用以及離子的穩定性。當酰胺轉化為磺酰胺時,N原子左側的酰基就變成了磺酰基。強的端基效應,意味著Z的離去能力更好。與N原子直接相連的酰基換成了吸電子能力更強的磺酰基后,就會與基團Z之間發生更強的吸電子競爭作用,從而削弱了端基效應。
[10-3] 本題考察加成-消除反應、自由基反應與氮賓的基本知識。tBuCOO-的離去能力強于BnO-,可確定tBuCOO-相當于[10-2]酰胺B中的離去基團Z。在-OBn孤對電子的參與下,通過給電子對的轉移,tBuCOO-基團離去,發生消除反應。底物二級胺的N原子提供孤對電子,親核進攻酰胺N原子,N=O間的一對π電子向O原子偏移,得到氮正離子中間體M6。氫質子被攫取后,受中間體M7內連著亞甲基的N的孤對電子影響,BnO-基團離去,產生中間體M8。接下來BnO-重新進攻帶正電的酰基碳,經加成-消除反應后,得到二氮烯中間體M9。C-N鍵斷裂的同時,釋放出N2物種,生成了兩個相同的含Br自由基,兩者迅速結合形成產物。反應的機理如圖8所示,轉化過程中必定經過的兩個關鍵中間體分別為M7和M9。
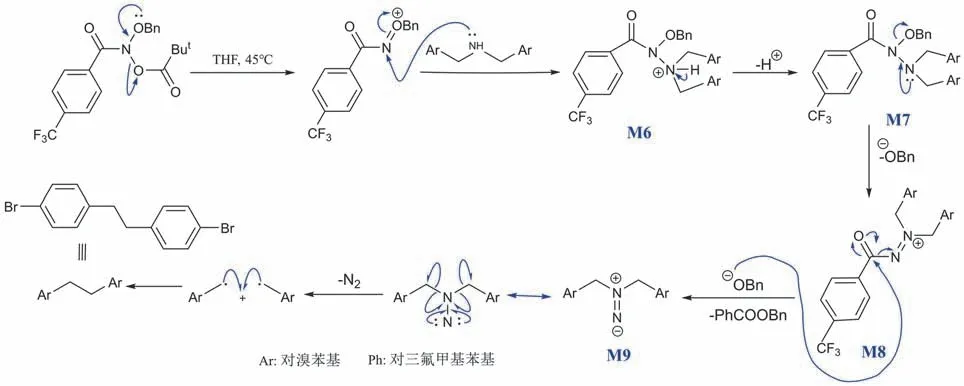
圖8 具有端基效應的酰胺對二級胺骨架進行編輯以構建C-C鍵的反應機理
2.2 評注與拓展
[10-2-1] 用分子軌道理論(MO)解釋本題,關鍵點是n(Y)和σ*(N-Z)兩個軌道間的相互作用[18]。從N、O到F,隨著Z原子的電負性依次增加,σ*(N-Z)軌道的能量逐漸降低,更加接近n(Y)軌道的能量;此外σ*(N-Z)軌道中N的成分逐漸增加,n(Y)與σ*(N-Z)軌道重疊的程度越大,體現了端基效應的調控作用,促進了化學反應的發生。端基效應(異頭效應)屬于靜態立體電子效應。關于立體電子效應,可簡要概括為:分子內的成鍵或非鍵電子在空間的特定方向產生軌道相互作用,進而引起分子的結構(含鍵長和鍵角)、能量與反應活性的變化[19]。這種效應可用于構象分析、立體化學以及反應過渡態的研究,是科學家在設計有機合成路線時不容忽視的因素之一[20]。
[10-2-2] 本題可從離子穩定性的角度考慮:基團Z離去后,N原子帶正電,左側酰基的吸電子作用不利于陽離子的穩定存在。當吸電子能力更強的磺酰基取代酰基后,形成的陽離子穩定性更差,基團Z就更難以離去,端基效應減弱。用MO來看:強吸電子基團可使N-Z發生極性反轉,由此改變N對σ*(N-Z)軌道的貢獻度。轉化為磺酰胺后,N原子帶δ-,Z原子帶δ+,σ*(N-Z)軌道中N的成分下降,n(Y)與σ*(N-Z)軌道的重疊性變小,不利于化學反應的進行。
[10-3] 題目選自2021年5月發表于Nature的一項研究成果[21]。C-H鍵官能化反應雖然發展迅速,但能直接用于潛在分子骨架修飾的案例不多,骨架轉化仍是一類尚未完全開發但發展潛力巨大的新型化學反應。從簡化逆合成步驟的角度出發,實現對單原子“插入”與“刪除”的操縱相當誘人。芝加哥大學化學系的Levin課題組發展了一種新型的分子編輯反應:直接“刪除”仲胺中的氮原子,借助異二氮烯中間體釋放出N2,實現分子內偶聯以構建C-C鍵。反應的關鍵在于某種異頭酰胺試劑可促進脂肪族仲胺的分子間活化。分子編輯技術有助于開發真正意義上的“無痕”化學反應——對合成有幫助的分子特征不殘留在產物中,為有機分子結構的優化提供了新的思路[22]。
3 第10題解析B
3.1 考題解析
[10-4] 本題考察Dyotropic重排反應在復雜分子合成中的作用。
[10-4-1] 該反應的驅動力是通過擴環釋放四元環的角張力。一個內酯環在擴張的同時,伴隨著另一環的縮合。C(4)與C(5)分別連接的酰氧基同時處于反式共平面,具備協同遷移的傾向。理論計算表明這種Dyotropic重排由TMS+基團引發,類似于雙重SN2反應[23-25],見圖9。

圖9 雙內酯通過雙重重排反應合成螺內酯
[10-4-2] 反應的驅動力仍然是四元環張力的釋放,故C(6)-C(7)右側遷移的是酰氧基。至于左側遷移的是σ(C-H)還是σ(C-C),可從以下角度思考:① 若遷移σ(C-H),形成的是六元環。若遷移σ(C-C),則形成七元環,七元環結構的穩定性低于六元環;② 遷移σ(C-C)要求C(6)-C(7)左側的烷基與右側的酰氧基同時處于反式共平面,而這種構象的基團斥力作用大,穩定性差,因此在整個構象體系中所占的比例低。相應地,氫與酰氧基在紙面上處于反式的構象穩定性好;③分析過渡態TS-3和TS-4的構象[26],見圖10。從空間效應的角度,TS-3的位阻相對較小,反應沿路徑a進行的可能性大。但DFT(密度泛函理論)計算表明:TS-3與TS-4兩種構象的能量較為接近。由于反應物具有構象靈活性,故空間效應不是唯一的決定因素。再者,σ(C-H)鍵軌道的能量通常比σ(C-C)鍵軌道能量高,且TS-4中σ(C-H)與σ*(C-O)之間軌道重疊的匹配性更好。綜合上述因素,σ(C-H)鍵更傾向于遷移。反應的歷程如圖11所示。

圖10 C-C鍵與C-H鍵遷移的化學選擇性
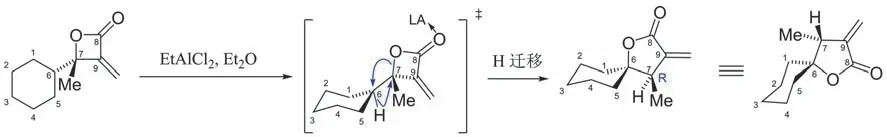
圖11 α-亞甲基-β-內酯的雙重重排反應
[10-4-3] 鋰代二氫呋喃與二甲基銅鋰(Gilman試劑)之間轉金屬化,在呋喃環上引入含Cu基團,構建了C-Cu鍵的分子骨架[27,28]。高階銅酸鹽中間體M10經過環狀過渡態,Cu上的甲基和烷氧基團相互遷移形成中間體M11,水解后得到產物。重排的驅動力可能為“強堿制備弱堿”,反應歷程見圖12。

圖12 有機銅試劑參與的雙重重排反應
[10-4-4] 含Pd基團與苯基之間相互遷移,生成的中間體在金屬Pd(II)的作用下還原消除直接得到非對映選擇性產物,重新構建了C-F鍵。重排的驅動力可能為:① 空間相互作用的弱化;② 生成了熱力學更穩定的α-羰基Pd(IV)物種[29]。反應的歷程見圖13。

圖13 金屬Pd基團參與的雙重重排反應
3.2 評注與拓展
[10-4-2] 選自2020年12月發表在AngewandteChemieInternationalEdition的一項研究成果[26]。清華大學的唐葉峰課題組借助環張力驅動的Dyotropic重排反應,構建了一種合成α-亞甲基-g-丁內酯的通用方法。反應中遷移基團的遷移順序具有一定的規律性,可實現產物的取代類型和立體化學的精準調控。研究指出該反應具有廣泛的底物適用性,已被成功應用于一些天然產物及其核心骨架、生物探針分子等的合成,在某種程度上解決了潛在的副反應易發生、化學選擇性難受控的問題,凸顯了方法的實用性。
[10-4-3]與[10-4-4]選自NatureChemistry在2021年7月刊出的一篇文章[29]。洛桑聯邦理工學院的祝介平團隊成功實現了在溫和的條件下鄰位C-C鍵和C-Pd(IV)鍵的Dyotropic重排,并構建了一系列含有三個立體中心的氟化環戊烷,反應具有高度的立體專一性。這種重排過程為常規手段難以實現的C-C鍵活化創造了新的開端,且有望作為Pd催化反應的一種工具。
4 結語
第35屆中國化學奧林匹克(初賽)有機化學部分的試題難度與往屆相比變化不大,但立體化學的復雜性提升,涵蓋的知識面更廣,對選手的綜合能力與高階思維提出了進一步的要求。學生在日常訓練中可參考國內外的著名期刊或高等有機化學書籍,加強反應機理的理解與推導。此外,鼓勵學生接觸化學發展的前沿,拓寬自身的視野,培養分析、運用信息解決綜合問題的能力與創新型思維。
猜你喜歡
小學科學(學生版)(2021年3期)2021-04-13 08:26:20
科技知識動漫(2017年7期)2017-08-09 19:52:45
科技知識動漫(2017年5期)2017-05-11 21:34:16
科技知識動漫(2017年4期)2017-04-15 22:24:55
科技知識動漫(2017年2期)2017-02-06 20:59:46
科技知識動漫(2016年10期)2016-10-18 20:35:00
中學生天地(C版)(2016年2期)2016-08-23 12:06:30
考試周刊(2016年63期)2016-08-15 22:51:06
中學生數理化·中考版(2015年12期)2015-09-10 07:22:44
發明與創新(2015年25期)2015-02-27 10:39:25
