加味星蔞承氣顆粒制備工藝及質(zhì)量標(biāo)準(zhǔn)研究
2023-08-09 14:33:42任桂林蒲清榮趙劍黃銳羅群唐定洪李能源楊思進(jìn)
云南中醫(yī)中藥雜志 2023年7期
任桂林 蒲清榮 趙劍 黃銳 羅群 唐定洪 李能源 楊思進(jìn)

摘要:目的 研究加味星蔞承氣處方的制備工藝和質(zhì)量標(biāo)準(zhǔn),為研制成顆粒劑提供基礎(chǔ)。
方法 采用正交實(shí)驗(yàn)法進(jìn)行水提工藝研究,以出膏率及辛弗林的含量為指標(biāo),優(yōu)選出最佳的水提工藝。采用薄層鑒別及高效液相色譜法對加味星蔞承氣顆粒進(jìn)行質(zhì)量標(biāo)準(zhǔn)研究。
結(jié)果 加味星蔞承氣處方最佳的提取工藝為:十二倍量的水,浸泡0.5 h,煎煮0.5 h,煎煮3次。在此條件下,出膏率為(21.79±0.94)%,辛弗林含量為(2.0097±0.09)mg/g。該水提工藝條件穩(wěn)定可行,干膏率及辛弗林含量較高。加味星蔞承氣顆粒以羌活中紫花前胡苷,大黃中大黃酸、大黃素甲醚,芒硝為鑒別指標(biāo),以麩炒枳實(shí)中辛弗林為含量指標(biāo),建立了加味星蔞承氣顆粒的質(zhì)量標(biāo)準(zhǔn)。
結(jié)論 本文為加味星蔞承氣顆粒備案為醫(yī)療機(jī)構(gòu)制劑提供了藥學(xué)基礎(chǔ)。
關(guān)鍵詞:加味星蔞承氣顆粒;制備工藝;質(zhì)量標(biāo)準(zhǔn);辛弗林;正交實(shí)驗(yàn)
中圖分類號:R285.5
文獻(xiàn)標(biāo)志碼:A
文章編號:1007-2349(2023)07-0001-03
痰熱腑實(shí)證是中風(fēng)病急性期最常見的證候,化痰通腑法是中風(fēng)病急性期基本治療大法[1]。星蔞承氣湯是化痰通腑的代表方劑[2]。加味星蔞承氣湯是在星蔞承氣湯的基礎(chǔ)上加郁金、石菖蒲、膽南星、雞血藤、麩炒枳實(shí)、姜厚樸、地龍、甘草組成,具有熄風(fēng)清熱,通腑化濁,開玄啟閉的功效,適用于中風(fēng)病急性期風(fēng)火痰瘀所致的玄府郁閉證,癥見神識昏朦、口舌歪斜、偏癱、肢麻、大便秘結(jié)或不通等。為方便臨床使用,提高治療腦梗死急性期治療水平,本文采用正交實(shí)驗(yàn)優(yōu)選加味星蔞承氣處方最佳的制備工藝,并研制成顆粒劑,同時對顆粒劑進(jìn)行質(zhì)量標(biāo)準(zhǔn)研究,為醫(yī)療機(jī)構(gòu)制劑的申報提供藥學(xué)基礎(chǔ)。
1 材料與方法
1.1 實(shí)驗(yàn)材料 紫花前胡苷(中國食品藥品檢定研究院,批號:111821-201604);辛弗林(中國食品藥品檢定研究院,批號:110727-201809);大黃酸(中國食品藥品檢定研究院,批號:110757-201607);大黃素甲醚(中國食品藥品檢定研究院,批號:110758-201817);加味星蔞承氣顆粒(西南醫(yī)科大學(xué)附屬中醫(yī)醫(yī)院制劑室,批號:20201128,20201129,20201130)
1.2 實(shí)驗(yàn)儀器 A-SERIES電子天平(常州市幸運(yùn)電子設(shè)備有限公司);AUW120D十萬分之一電子天平(日本島津);HH-4數(shù)顯恒溫水浴鍋(江蘇金壇市金城國勝實(shí)驗(yàn)儀器廠);JPCT0328超聲波發(fā)生器(武漢嘉鵬電子有限公司);GOODLOOK-1000型薄層成像系統(tǒng)(上海科哲生化科技有限公司);LC-20AT高效液相色譜儀(日本島津)
1.3 辛弗林含量測定[3] 色譜條件 以十八烷基鍵合硅膠為填充劑;以甲醇-磷酸二氫鉀溶液(取磷酸二氫鉀0.6 g,十二烷基磺酸鈉1.0 g,冰醋酸1 mL,加水溶解并稀釋至1000 mL)(50:50)為流動相;流速為1 mL·min-1,檢測波長為275nm,柱溫為40℃,理論塔板數(shù)按辛弗林計應(yīng)不低于4000。
對照品溶液的制備 取辛弗林對照品適量,精密稱定,置棕色量瓶中,加甲醇制成每1 mL含辛弗林0.5 mg的溶液,即得。
供試品溶液的制備 取干浸膏粉末3 g,精密稱定,置具塞錐形瓶中,精密加入甲醇50 mL,稱定重量,加熱回流1.5 h,放冷,再稱定重量,用甲醇補(bǔ)足減失的重量,搖勻,濾過,精密量取續(xù)濾液10 mL,蒸干,殘?jiān)铀?0 mL使溶解,通過聚酰胺柱(60~90目,2.5 g,內(nèi)徑為1.5 cm,干法裝柱)用水20 mL洗脫,收集洗脫液,轉(zhuǎn)移至25 mL量瓶中,加水至刻度,搖勻,即得。
1.4 制備工藝研究 按處方準(zhǔn)確稱取加味星蔞承氣飲片,選擇加水量、浸泡時間、煎煮時間、煎煮次數(shù)為因素,選用L9(34)因素水平表安排正交試驗(yàn),進(jìn)行水提,合并提取液,濃縮,烘干,進(jìn)行干膏率的測定及含量測定,優(yōu)選最優(yōu)的水提工藝[4-5]。
1.5 質(zhì)量標(biāo)準(zhǔn)研究 對加味星蔞承氣顆粒進(jìn)行質(zhì)量標(biāo)準(zhǔn)研究,包括鑒別和含量測定。以羌活中紫花前胡苷、大黃中的大黃酸、大黃素甲醚和芒硝作為鑒別,以枳實(shí)中的辛弗林作為含量測定指標(biāo)。
2 結(jié)果
2.1 方法學(xué)考察[6]
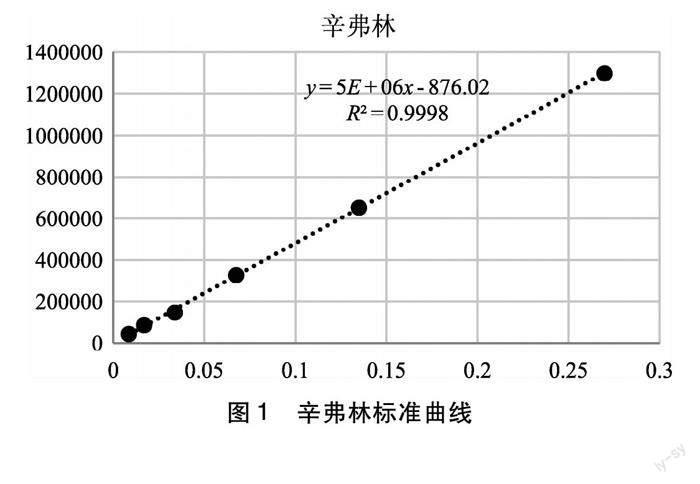
2.1.1 線性關(guān)系的考察 取辛弗林對照品8.47 mg置25 mL量瓶中,加甲醇使溶解并稀釋至刻度,搖勻,制得濃度為0.3371 mg/mL的對照品溶液,作為對照品貯備液,備用。分別精密量取對照品貯備液0.25,0.5,1.0,2.0,4.0,8.0 mL,分別置10 mL的量瓶中,加甲醇至刻度,搖勻,分別制成濃度為0.008 43,0.016 86,0.033 71,0.067 42,0.134 84,0.269 68 mg/mL的對照品溶液。精密吸取上述對照品溶液各10 μL,按照上述色譜條件測定。以峰面積(A)為縱坐標(biāo)(Y),辛弗林濃度(mg/mL)為橫坐標(biāo)(X),繪制標(biāo)準(zhǔn)曲線。得到辛弗林的回歸方程為y=5E+06x-876.02,R2=0.999 8(n=6),在0.008 43 mg/mL~0.269 68 mg/mL范圍內(nèi)呈良好的線性關(guān)系。
2.1.2 精密度試驗(yàn) 取濃度為0.033 371 mg/mL的辛弗林對照品連續(xù)進(jìn)樣測定6次,其峰面積平均值為148 364.3,RSD值為0.93%,表明儀器精密度良好。
2.1.3 重復(fù)性試驗(yàn) 取加味星蔞承氣浸膏粉,按供試品制備方法制備,平行制備6份樣品,測定,辛弗林平均含量為1.66 mg/g,RSD%為1.32%,表明分析方法重復(fù)性良好。
2.1.4 穩(wěn)定性試驗(yàn) 取同一供試品溶液,分別在0,2,4,8,12,24 h進(jìn)樣分析。記錄峰面積,結(jié)果辛弗林峰面積均值為206 886.5,RSD%為0.69%。表明供試品溶液在24 h內(nèi)穩(wěn)定。
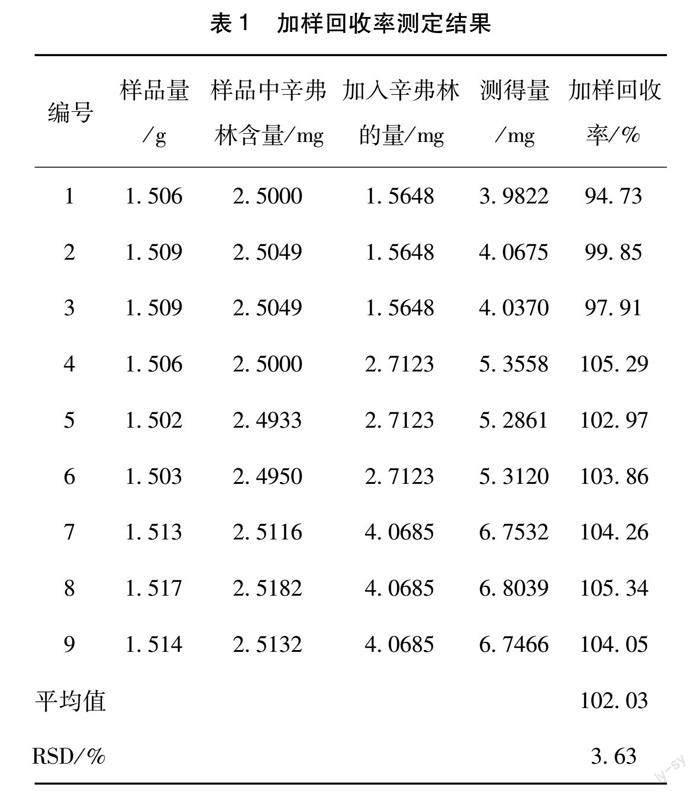
2.1.5 加樣回收試驗(yàn) 精密稱取辛弗林對照品26.21 mg,加甲醇定容至25 mL,其濃度為1.0432 mg/mL,稱取同一份供試品1.5 g,精密稱定。每3份為一組,每組分別加入辛弗林對照品溶液1.5 mL,2.6 mL,3.9 mL。按照供試品溶液制備方法制備,進(jìn)樣,測定。
2.1.6 專屬性試驗(yàn) 取缺枳實(shí)的處方量飲片,按“1.3辛弗林含量測定”中供試品制備方法制備陰性樣品溶液,對比陰性樣品、對照品和供試品的高效液相色譜圖。結(jié)果表明加味星蔞承氣顆粒其他組分對辛弗林的測定無干擾,本方法專屬性強(qiáng)。
2.2 制備工藝研究
2.2.1 水提正交實(shí)驗(yàn)研究 取水提正交實(shí)驗(yàn)干浸膏,按照辛弗林含量測定的方法制備樣品,測定,記錄峰面積,計算含量。以辛弗林的含量及干膏率為指標(biāo)進(jìn)行綜合評分,綜合評分值=[(辛弗林含量/辛弗林含量最大值)×0.6+干膏率/干膏率最大值×0.4]×100[7-8]。正交試驗(yàn)結(jié)果見表3、表4。
由直觀分析可見,影響提取效果的因素順序?yàn)椋禾崛〈螖?shù)C>加水量A>煎煮時間B>浸泡時間D,以浸泡時間D作為誤差項(xiàng),進(jìn)行方差分析。方差分析結(jié)果表明,提取次數(shù)和加水量對結(jié)果有顯著性影響(P<0.05),煎煮時間對結(jié)果無顯著性影響,無統(tǒng)計學(xué)差異(P>0.05)。因此,水提的最佳提取工藝中,最佳的因素組合為A3B1C3D3,考慮到生產(chǎn)實(shí)際,由于浸泡時間對結(jié)果影響很小,故最佳的水提工藝定為:十二倍量的水,浸泡0.5 h,煎煮0.5 h,煎煮3次。
2.2.2 水提驗(yàn)證性試驗(yàn) 由于水提工藝的最佳方案沒有在正交試驗(yàn)因素表內(nèi),因此對最佳水提工藝進(jìn)行了3次驗(yàn)證。結(jié)果見表5。
2.3 質(zhì)量標(biāo)準(zhǔn)
2.3.1 羌活TLC鑒別[9-10] 取加味星蔞承氣顆粒??? 5 g,加甲醇50 mL,超聲處理20 min,靜置,過濾,濾液濃縮至2 mL,作為加味星蔞承氣顆粒供試品溶液。另取紫花前胡苷對照品,加甲醇制成每1 mL含0.5 mg的溶液,作為對照品溶液。按處方(除羌活外)與制法,制成陰性樣品,其余處理同供試品溶液,作為陰性樣品溶液。照薄層色譜法(四部通則0502)試驗(yàn),吸取上述三種溶液各10 μL,分別點(diǎn)于同一3%醋酸鈉溶液制備的硅膠G薄層板上,以三氯甲烷-甲醇(8∶2)為展開劑,展開,取出,晾干,置紫外光燈(365 nm)下檢視。供試品色譜中,在與對照品色譜相應(yīng)的位置上,顯相同的藍(lán)色熒光斑點(diǎn),且陰性樣品無干擾。同時在三氯甲烷-乙酸乙酯-甲醇-甲酸(9∶1∶5∶0.5)驗(yàn)證系統(tǒng)一和三氯甲烷-甲醇(9∶5)驗(yàn)證系統(tǒng)二為展開劑的條件下,樣品中分離情況均良好,缺羌活陰性樣品無干擾。見圖3。
2.3.2 大黃TLC鑒別[11-14] 取加味星蔞承氣顆粒5 g,加甲醇50 mL,浸泡1 h,濾過,蒸干,殘?jiān)铀?0 mL使溶解,再加鹽酸1 mL,加熱回流30 min,立即冷卻,用乙醚分2次振搖提取,每次20 mL,合并乙醚液,蒸干,殘?jiān)尤燃淄? mL使溶解,作為加味星蔞承氣顆粒供試品溶液。另取大黃酸和大黃素甲醚對照品,分別加甲醇制成每1 mL含1 mg的溶液,作為對照品溶液。按處方(除大黃外)與制法,制成陰性樣品,其余處理同供試品溶液,作為陰性樣品溶液。照薄層色譜法試驗(yàn),吸取上述四種溶液各10 μL,分別點(diǎn)于同一以羧甲基纖維素鈉為黏合劑的硅膠H薄層板上,以石油醚(30~60℃)-甲酸乙酯-甲酸(15∶5∶1)的上層溶液為展開劑,展開,取出,晾干,置紫外光燈(365 nm)下檢視。供試品色譜中,在與對照品色譜相對應(yīng)的位置上,顯相同的橙黃色熒光主斑點(diǎn),陰性無干擾。同時在正己烷-乙酸乙酯-甲酸(30∶10∶0.5)驗(yàn)證系統(tǒng)一和環(huán)己烷-乙酸乙酯-甲酸(12∶5∶0.1)驗(yàn)證系統(tǒng)二為展開劑的條件下,樣品中分離情況均良好,缺大黃陰性樣品無干擾。見圖4。
2.3.3 芒硝鑒別 芒硝中含有Na2SO4,取加味星蔞承氣顆粒1 g,加10 mL水溶解后,加鹽酸1 mL,攪拌,加入氯化鋇試液5 mL,作為加味星蔞承氣顆粒供試品溶液。另取芒硝0.2 g,加10 mL水溶解后,加鹽酸1 mL,攪拌,加入氯化鋇試液5 mL,作為芒硝對照溶液。芒硝對照溶液和加味星蔞承氣顆粒供試品均生成白色沉淀。
2.3.4 含量測定 加味星蔞承氣顆粒的含量測定色譜條件及供試品制備方法同“1.3辛弗林含量測定”,根據(jù)加味星蔞承氣顆粒(批號:20201128,20201129,20201130)三批中試結(jié)果,規(guī)定本品每1 g顆粒含麩炒枳實(shí)以辛弗林(C9H13NO2)計,不得少于0.94 mg。
3 討論
加味星蔞承氣顆粒供試品色譜中,在與紫花前胡苷相對應(yīng)的位置上,顯相同顏色的藍(lán)色熒光斑點(diǎn),且陰性無干擾,可作為質(zhì)量標(biāo)準(zhǔn)鑒別項(xiàng)。加味星蔞承氣顆粒供試品色譜中,在與大黃酸、大黃素甲醚相對應(yīng)的位置上,顯相同顏色的橙黃色熒光斑點(diǎn),且陰性無干擾,可作為質(zhì)量標(biāo)準(zhǔn)鑒別項(xiàng)。加味星蔞承氣顆粒供試品與芒硝顯相同的白色沉淀,可作為質(zhì)量標(biāo)準(zhǔn)鑒別項(xiàng)。
同時,本文對其他部分藥材也進(jìn)行了鑒定試驗(yàn),加味星蔞承氣顆粒供試品色譜中,在與郁金、姜厚樸、地龍對照藥材相應(yīng)的位置上,無相應(yīng)的斑點(diǎn),故未選擇郁金、姜厚樸、地龍作為鑒別。加味星蔞承氣顆粒供試品色譜中,在與瓜蔞皮對照藥材相應(yīng)的位置上,斑點(diǎn)顏色較淺,加大取樣量和點(diǎn)樣量后,斑點(diǎn)顏色仍較淺,故不作為質(zhì)量標(biāo)準(zhǔn)的鑒別指標(biāo)項(xiàng)。加味星蔞承氣顆粒供試品色譜中,在與雞血藤對照藥材相應(yīng)的色譜位置上,紅色主斑點(diǎn)無相應(yīng)斑點(diǎn),故未選擇雞血藤作為鑒別項(xiàng)。加味星蔞承氣顆粒和缺膽南星陰性樣品在兩液相交處均能產(chǎn)生環(huán),故膽南星不作為鑒別項(xiàng)。加味星蔞承氣顆粒中含量測定以辛弗林為指標(biāo),且缺麩炒枳實(shí)陰性樣品無干擾,可作為含量測定指標(biāo)。
本文按照四川省醫(yī)療機(jī)構(gòu)機(jī)制備案管理的相關(guān)
要求,本文主要完成了工藝研究和質(zhì)量標(biāo)準(zhǔn)研究,為醫(yī)療機(jī)構(gòu)制劑的申報提供基礎(chǔ)。
參考文獻(xiàn):
[1]劉宏偉,王一戰(zhàn),鄒憶懷.星蔞承氣湯治療急性缺血性腦卒中的效應(yīng)機(jī)制研究現(xiàn)況[J].中國中醫(yī)急癥,2022,31(7):1294-1296.
[2]張旭紅.星蔞承氣湯治療急性腦梗死痰熱腑實(shí)證患者的臨床療效[J].臨床合理用藥雜志,2022,15(6):17-19.
[3]國家藥典委員會.中國藥典[M].北京:中國醫(yī)藥科技出版社,2020:258.
[4]黃慶寶,梁慧超.正交實(shí)驗(yàn)設(shè)計法優(yōu)選復(fù)方腎康顆粒水提工藝[J].湖北中醫(yī)雜志,2020,42(2):52-54.
[5]黃慶寶,黃詩瑩,陳劍平,等.正交試驗(yàn)設(shè)計法優(yōu)選六高康顆粒水提工藝[J].中國藥業(yè),2022,29(7):74-77.
[6]耿彥梅,高晗,李雪利,等.枳實(shí)標(biāo)準(zhǔn)湯劑質(zhì)量研究[J].河北工業(yè)科技,2021,38(1):63-70.
[7]黃銳,楊思進(jìn),蒲清榮,等.正交試驗(yàn)優(yōu)選清肺排毒合劑(新冠1號)制備工藝[J].云南中醫(yī)中藥雜志,2020,41(5):5-7.
[8]謝月,梁奇,馮育林,等.參芪益氣消渴丸的水提與制劑工藝的優(yōu)化[J].江西中醫(yī)藥,2020,51(6):70-74.
[9]孫莉.馬氏接骨膏薄層色譜鑒別[J].海峽藥學(xué),2020,32(4):53-55.
[10]施敏,管敏.上感合劑質(zhì)量標(biāo)準(zhǔn)研究[J].中國藥業(yè),2019,28(3):42-44.
[11]張玉娥,王賢兒,關(guān)琴笑,等.散瘀止痛凝膠膏劑質(zhì)量標(biāo)準(zhǔn)研究[J].亞太傳統(tǒng)醫(yī)藥,2019,15(3):54-56.
[12]王暢,張蕊多,王仁廣,等.榛苓顆粒質(zhì)量標(biāo)準(zhǔn)研究[J].實(shí)用藥物與臨床,2017,20(11):1314-1318.
[13]溫馨,江耀倫,陳素珍,等.大七厘散中大黃及當(dāng)歸薄層色譜鑒別方法改進(jìn)[J].中國藥業(yè),2017,26(14):7-9.
[14]袁航,謝寒冰,陳安珍,等.清火片標(biāo)準(zhǔn)中檢測方法的提高與修訂研究[J].中國藥品標(biāo)準(zhǔn),2019,20(1):51-56.
(收稿日期:2022-08-18)
