骨骼肌衰減癥與ATM 蛋白缺失模型的比較研究
——運動抗衰老模型
2023-07-31 09:00:10王雅薇張瓊月
湖北體育科技 2023年7期
金 晶,王雅薇,張瓊月,葉 蕾
(1.杭州師范大學體育學院,浙江 杭州 310000;2.浙江農林大學動物科技學院,浙江 杭州 311300;3.浙江農林大學經管學院,浙江 杭州 311300;4.杭州師范大學數學學院,浙江 杭州 310000)
物質資源和醫療水平的飛躍式發展后“人口老齡化”的進程在全世界不斷推進,尤其是擁有龐大人口的中國,我國人口年齡結構已經出現“反差式”的轉變[1]。 伴隨著年齡遞增,骨骼肌衰減癥也隨之發生,引起老年人起坐困難、日常活動受限、運動能力減弱,甚至摔倒的風險,嚴重困擾老年人自理能力和生活質量。 衰老內在機制和抗衰老方法的探究已有一代又一代的科學工作者持續研究熱點, 因此骨骼肌衰減癥(Sarcopenia)相關研究受到專家、學者的青睞[2-3]。
ATM 小鼠模型作為衰老的模型已有廣泛研究[4-5],由于其生長停滯、骨骼肌萎縮等骨骼肌退行性變化的表征[6-7],可區別于其他的衰老動物模型。 因此本文擬通過對比ATM 蛋白缺失與骨骼肌衰減癥的診斷依據、 老年疾病與內在發病機制等方面,探索ATM 動物模型作為骨骼肌衰減癥模型的可能性。
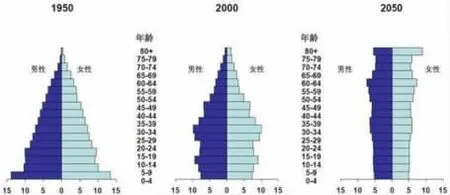
圖1 中國人口年齡結構的預測[1]
1 骨骼肌衰減癥診斷共識指標比較
與骨質疏松病癥的經歷相類似, 骨骼肌衰減癥的發展同樣經過了“從無到有”的過程,從最開始的“表征”的提出,到之后的“病癥”的確立。 歐盟骨骼肌衰減癥工作小組在2010 年建立了評估指標,分別從肌肉質量、力量和身體活動能力3 個方面進行細化和深化[8]。亞洲關于這方面的研究稍晚,于2013 年成立骨骼肌衰減工作組, 也采用了與歐盟雷同的策略, 并在2014 年發表了亞洲肌少癥診斷共識也包括肌肉質量、 肌肉力量和肌肉功能3 個方面[9],因此本文也從這3 個方面進行比較和討論。
1.1 Sarcopenia 與AT 病人
1.1.1 骨骼肌質量與肌力
世界范圍內“老齡化問題”日益突出,Sarcopenia 已成為高發態勢。 有2 項獨立對不同人群進行統計研究表明:人體骨骼肌質量在20~90 歲之間大約下降了50%,骨骼肌力量在30 歲時達到峰值, 并且達到40 歲以后骨骼肌質量下降8%/10 年,70 歲以后大約下降15%/10 年[10];0~75 歲的165 位受試者的研究顯示,骨骼肌質量隨年齡增加而減少,尤其是II 型肌纖維質量和數量[11]。 與正常同齡對照組相比,AT 患者的體型矮小,生長停滯,身高、體重及BMI 值顯著偏低:Ruiz-Botero 報告的14 歲女孩身高僅為1.38 m、體重27 kg[12];以色列國家實驗室的52 名病人[13]、澳大利亞研究團隊對25 名患者[14]以及英國70 人[15]的研究都發現體重下降現象,有研究報告對AT 病人的尸檢發現患者的骨骼肌呈現不同程度的萎縮[16]。 通過體成分的顯示,衰老會導致骨骼肌II 型肌纖維下降,而脂質(脂肪)增多[17]和I 型肌纖維無變化[18],認為AT 病人的組織中脂類和脂肪組織并未改變, 而體重下降的主要原因是由于骨骼肌質量的下降[14],因此提示衰老和AT 病人都出現骨骼肌質量下降現狀。
骨骼肌的力量伴隨著增齡性的改變, 肌肉力量隨年齡逐年下降。AT 病人的力量測試研究包括呼吸肌測定[19]和握力測試[14],均顯示顯著低于正常對照組,表明ATM 蛋白缺失將影響個體的骨骼肌力。
1.1.2 運動能力/身體活動能力
衰老引起骨骼肌質量和肌力的不斷下降,隨之產生骨骼肌衰減癥高發, 持續加劇的肌力下降將破壞現有的生活方式,更為嚴重的將致使增加骨骼肌的損傷幾率甚至發生跌倒概率[3],嚴重影響老年人的自理能力[2]。 Snijders[17]研究團隊分析青年組和老年組的生理指標,BMI 老年組顯著高于青年組,但是腿部骨骼肌質量卻顯著偏低, 而且最大腿部伸展和收縮力量均顯著低于青年組,亦說明衰老導致骨骼肌質量下降,降低機體的運動能力。 AT 病人從2~3 歲開始需要依靠外力進行站立,5~10 歲逐步失去行走能力。 個案研究發現6 歲出現遞進式的運動失調和四肢顫抖,7 歲時完全依賴于外界控制站立[12];Pommerening[14]等對25 名研究對象進行分析,將能否獨自站立分為行走組和輪椅組,結果顯示10 歲以上患者完全依賴于輪椅活動;Woods[15]統計認為AT 病人平均8 歲將失去書寫能力不得不停止教育,也佐證10 歲將失去行走能力。
1.2 動物模型及組織的對比
權威文獻Nature 上發表關于成年鼠(5~6 月齡)、衰老鼠(20~24 月齡)、老態鼠(28~32 月齡)之間骨骼肌肌纖維數量和肌纖維尺寸的研究, 肌纖維數量結果顯示成年鼠>衰老鼠>老態鼠(p<0.05);肌纖維橫截面積顯示成年鼠>衰老鼠>老態鼠(p<0.05)[20]。 衰老小鼠骨骼肌肌力和肌量的下降,研究認為與蛋白質合成通路受到抑制相關[21],也有研究指出細胞外基質的變化引起機械性能的變化[22-23]。 研究報道運動干預后動物模型的有益積累效果減弱或無變化, 進而表明衰老將導致小鼠運動能力逐步削弱[24]。
ATM-/-小鼠與野生型小鼠相比體型更小,Ching[25]等發現純合子的體重是野生型小鼠的70%左右 (16.7±1.2 g vs 23.7±2.2 g,p<0.01);Quek[26-27]等在構建的ATM 大鼠敲除模型顯示,實驗動物出現下肢癱瘓和骨骼肌萎縮現象。 ATM 動物模型發現旋轉臺測試性能下降, 而且步長較短和步型不一致的現象出現,還發生輕度運動學習障礙[28-29]。
1.3 改善癥狀的方法探究
1.3.1 運動干預比較
運動可改善骨骼肌衰減癥表征, 加強骨骼肌肌力及骨骼肌的增粗肥大,可能通過調控骨骼肌中的細胞外基質、氧化還原狀態、細胞自噬等信號通路[30]。 中等強度的有氧運動有改善作用:人體實驗包括下坡跑訓練[31](坡度為-10°,85%最大攝氧量,12×5 min/次)、自行車蹬車實驗[32](6 周,4 次/周,30 min/次)和12 周的有氧自行車訓練[33](45 min/次,3 次/周,75%最大攝氧量),有效增加肌纖維的橫截面積;動物實驗包括SD 大鼠跑臺運動[34](17 m/min,90 min)、衰老小鼠跑臺運動[35](6 次/周,11.5 m/min,30 min/次)改善骨骼肌質量和衛星細胞數量。抗阻運動一直被認為是改善衰老骨骼肌質量和功能的關鍵手段:Snijders[11,23,36]團隊常年研究衰老機體通過抗阻運動后的變化,結果認為骨骼肌橫截面積和肌纖維增加, 有效改善骨骼肌功能以及增多衛星細胞含量;Dreyer[37]等獨立研究團隊也得到類似的結果,雖然較青年組效果有所下降。
盡管運動干預AT 病人的實例不多,但研究結果都具有良性效果:Felix[19]等設計24 周的呼吸肌訓練實驗,通過訓練后實驗組最大吸氣壓和最大呼氣壓顯著提升, 顯著緩解呼吸困難的感覺。 運動干預對2 種疾病模型均顯示有顯著性提升的效果,認為運動訓練有助于改善病狀。
1.3.2 藥物/營養干預比較
除了運動干預以外,藥物、營養和聯合干預(藥物/營養/運動相結合的方式)都能延緩/改善衰老個體的骨骼肌質量和功能的作用。 藥物治療骨骼肌衰減癥的有效靶點分子不斷的被發現,包括選擇性雄激素受體分子、胃饑餓素激動劑、肌生成抑制素抗體、激活素IIR 拮抗劑、血管緊張素轉換酶抑制劑、β拮抗劑、快速骨骼肌肌鈣蛋白激活劑等[38]。 動物[39]和人體[40]實驗通過氨基酸補給促進骨骼肌蛋白質合成增加; 通過維生素D 補給防治缺乏引起的骨質疏松、骨骼肌衰減等疾病[41];另外維生素C、E 和類胡蘿卜素等微量要素的補給有助于骨骼肌衰減癥[42]。 運動與營養/藥物的聯合干預有助于骨骼肌功能和質量的提升[43],認為3 個月內對提高步行速度有積極作用[44]。
AT 病人身高、體重及BMI 顯著較小,因而許多研究對AT病人營養狀況及生長激素進行分析,及時營養/藥物補給的研究早已實施但無效果。 例如有研究發現AT 病人6 歲缺乏生長激素,采用6 個月的生長激素治療,無法改變病人運動失調和顫抖情況[12];19 名病人存在營養不良現象,給予充足營養條件,BMI 仍然下降[45];2 項研究均發現AT 病人缺乏維生素D[13],且維生素D 的缺失與行走能力直接相關[14];給予病人維生素A和鋅充足補給,也無法改善AT 病人瘦體重偏低的現狀[46]。
通過2 種疾病受到藥物/營養的干預實驗得出, 藥物、營養和聯合干預對于衰老有顯著的改善作用,而對于AT 疾病無有益作用。

圖2 本研究疾病的骨骼肌衰減癥表現、疾病及其相關機制
2 衰老常見疾病與ATM 缺失表型比較
2.1 ATM 與阿爾茨海默癥
阿爾茨海默癥是一種增齡性神經系統疾病, 主要以記憶力減退和認知功能障礙為臨床特征, 并以老年斑和突觸丟失為主要病理特征的疾病, 常見于中老年人群也是癡呆癥的一種[50]。 近期,Kreis 研究團隊認為大腦發育蛋白(Drebrin,DBN)可調控神經發育過程中的細胞骨架的功能, 其作用與衰老、AD 疾病的結構和突觸功能變化相關[6]。Kommaddi 等證實DBN對氧化應激誘導的功能障礙具有保護機制,其機制為ATM 刺激DBN(ATM 的底物)調節細胞骨架動態的信號,抵御氧化應激和能量代謝的損傷,證實ATM 主要靶點是DBN 的647 號絲氨酸磷酸化,可改善AD 的早期癥狀[51]。 也有研究認為ATM-/-小鼠與AD 疾病具有一定的相似性作為AD 疾病模型,可通過定期給予布洛芬,減少神經炎癥反應、促進神經存活、增加嚙齒動物不同神經退行性的識別能力,其中包括AD、創傷性腦損傷和過度興奮性毒性等[52]。
AT 疾病一般發生在嬰幼兒時期(5 歲之前)導致多組織失調現象, 如核心中樞神經發生退行性的變化, 主要發生在小腦;而AD 大部分發生在生命的中后期(尤其是70 歲以后),海馬體、額葉皮質和皮質下的初級神經元的損傷為主,盡管2種疾病之間有部位、年齡等的差異,但是卻發現AD 患者的神經元死亡與ATM 信號消失相關[53]。
在人類AD 中, 組蛋白去乙酰化酶4 的核轉移、 組蛋白H3 的三甲基化以及細胞周期活性的降低,這3 個獨立角度都證實ATM 功能降低, 直接提示ATM 功能失調可能是AD 個體神經元死亡的一個重要原因。 為了更好的觀察AD 與ATM的關系,Shen 等采用3 種AD 嚙齒動物轉基因模型 (R1.40 小鼠APP 轉基因、PS/APP 小鼠和3xTG 小鼠;相對應的野生型作為對照組),無一例外都支持AD 大腦中ATM 功能的喪失;AD病人活檢細胞對ATM 標志物HDAC4 的檢測也發現ATM 信號顯著下降[54];研究還對AD 病人腦組織進行ATM 轉錄和翻譯情況進行檢測,結果一致認為ATM 的功能顯著下調[53]。
2.2 ATM 與癌癥
有研究認為ATM 蛋白缺失所引起的癌癥, 與DNA 雙鏈斷裂修復缺陷相關, 這種毒性應激加劇遺傳不穩定導致癌癥的發生[55],另一種觀點認為AT 病人顯示出免疫力下降,也會造成癌癥風險的升高[56]。 Choi 等對ATM 突變的區域解析,結果發現ATM 突變發生幾乎沒有重復的突變位置, 也就是說ATM 整個基因序列隨機的發生突變[57]。

表1 基于骨骼肌衰減癥診斷共識的比較
關于ATM 與癌癥的研究, 可追溯到19 世紀80 年代,AT病人可增加乳腺癌的發生率,隨后乳腺癌在家族遺傳中發現,之后越來越多的癌癥被發現與ATM 蛋白缺失相關[58]。 Helgason[59]等發現有許多基因直接通過ATM 進行磷酸化(BRCA1、BRCA2、TP53、PALB2、CHEK2 和NBN 等), 或依賴于ATM 功能進行誘導磷酸化作用發生。AT 病人擁有很高的患癌癥風險約25%, 而且在病人的前20 年發生淋巴瘤和白血病最為常見,年齡較大的病人則逐步發展成固態腫瘤包括乳腺癌和腸胃癌等[58],2011 年一項關于英國和荷蘭296 名AT 病人進行調查,其中有66 名患者確診為癌癥(47 名淋巴瘤和19 名非淋巴瘤),還證明兒童時期缺乏ATM 蛋白與淋巴樣腫瘤相關,發現有8 例乳腺癌病例,AT 患者乳腺癌風險增加了30 倍[60]。另一項研究對法國279 名AT 病人分析,69 名癌癥患者,包括8 名急性白血病(4 名T 細胞急性白血病)、12 名霍奇金淋巴瘤、38 名非霍奇金淋巴瘤、3 名發展為T 細胞前淋巴細胞白血病等[56]。
ATM 攜帶者(雜合型,即含有一條突變的ATM 基因個體)通常情況下表現為健康個體, 系統分析認為ATM 攜帶者得癌癥和心臟病的比率增加,且縮短壽命。雖然ATM 在普通人群中的存在率僅僅只有1%~2%, 但是雜合型的個體患癌癥的風險男生為2.3 倍,女生為3.1 倍[61],Jerzak[62]研究認為雜合型人群中終身發生乳腺癌的風險超過25%,因此建議40 歲以后要進行每月一次的自檢和每年一次的專項檢測。 ATM 被列為中度風險的乳腺癌標志基因,2016 年的meta 分析攜帶者人群發現乳腺癌發生率在50 歲時6% vs 80 歲時的30%[63]。Thompson[64]等也報道ATM 雜合型的乳腺癌分別存在中年組高于青年2倍以上,也存在女性高于男性的現象。 除了乳腺癌較為常見以外,文獻還報道了消化道癌癥、結腸直腸癌、胃癌等發生風險顯著提升[64-65]。
2.3 ATM 與糖尿病
早在20 世紀70 年代,Schalch 等發現一例特殊的AT 病人,顯示出以胰島素抵抗的糖尿病患者的癥狀,無獨有偶,AT病人餐后會發生血糖異常現象, 其中有一部分都患有糖尿病癥狀,通常出現在疾病進展的晚期[55,66],Morio 報告AT 病人有更高的風險發展成為糖尿病[67]。 近期,全基因相關性研究中發現單核苷酸多態位點 (SNP)rs11212617 與二甲雙胍血糖反應有關, 該位點正好位于11 號染色體的一個連鎖不平衡區域,共擁有7 個候選基因。ATM 基因恰好位于該區域中,雖然之前并沒有相關報道其與二甲雙胍有相互作用的關系,但已經有證據表明ATM 與葡萄糖代謝有相關性[55]。 早期體外實驗中觀察到ATM 與多種胰島素信號(JNK、PI3K、IRS2 和Akt)的相互作用關系[68],2019 年筆者針對ATM 與IGF1-Akt-mTOR-4EBP1通路的相關性進行探討,進行系統性的綜述,闡明ATM 直接或間接作用于各要素[69]。
另一個角度也證實ATM 缺失與血糖濃度上升有關。 動物實驗:Miles 通過對12 周齡小鼠進行糖耐受實驗,發現ATM-/-小鼠的血糖高于野生型,21~27 周齡時也發現相似的現象,胰島素的分泌量在ATM-/-小鼠骨骼肌中持續下降[70];通過氯喹的注射扭轉ATM 缺陷小鼠糖尿病癥狀, 增加胰島素刺激的Akt 的磷酸化作用[71]。 Connelly 等采用AT 患者和對照組進行口服葡萄糖耐量試驗,采用連續的血糖和胰島素測定,研究發現葡萄糖濃度分別為6.75 mmol/l、4.93 mmol/l(p=0.029),胰島素濃度分別為285.6 pmol/l、148.5 pmol/l(p=0.043),結論表示ATM 導致AT 病人的血糖升高和胰島素敏感性降低有關[72]。
2.4 ATM 與其他老年病
ATM 除了與上述疾病有相關研究以外, 還發現與亨廷頓病、心血管疾病、關節炎、骨質疏松等有關。 亨廷頓病,也叫做大舞蹈病或亨廷頓舞蹈癥,是常染色體顯性遺傳的疾病,該病是由于Huntington 基因發生變異, 產生了變異蛋白后逐步產生大的分子團影響神經細胞的功能。 HD 病人DNA 損傷時是由DNA 修復過程中Huntington 基因蛋白功能障礙引起的,因而活性氧的升高引起的DNA 氧化損傷加劇積累可能是HD發病的重要原因之一,Maiuri 的研究顯示Huntington 蛋白損傷位點依賴于ATM 蛋白的活性[73]。 Tidball[74]和Ferlazzo[75]的2項研究都認為ATM 缺失對HD 形成有關,HD 的修復和功能發揮依賴于ATM 蛋白,還認為錳離子的下降是引起ATM-p53信號通路阻斷, 導致ATM 下游靶點目標基因無法激活的原因。 Lu[76]的細胞實驗中得出相反的結論,研究發現HD 小鼠和HD 患者的腦組織中,ATM 信號持續升高, 雖然也認為ATM蛋白可能是HD 一個關鍵的治療靶點, 但是卻認為減少ATM信號有利于改善HD 癥狀。
ATM 與心血管疾病(冠狀動脈硬化/動脈粥樣硬化血管疾病)的研究主要是依賴于SNP 的相關性探索。德國通過240 名II 型糖尿病男性病人進行rs11212617 位點的研究,證明ATM依賴的信號可能對硬化血管疾病有重要作用[77];早期國內學者對562 名病人的rs189037 多態位點進行分析, 通過檢測ATM mRNA 表達發現該位點可能影響ATM mRNA 的表達量,進而對動脈硬化起作用[78];隨后針對國內漢族1 308 樣本(實驗組652 人和對照組656 人)的rs189037 進行探索,研究證實ATM 基因中rs189037 與中國漢族人群中冠狀動脈硬化有關,rs189037 位點的TT 表型有更低的心血管疾病可作為CAD 的遺傳學指標[79]。多態位點的結果雖然已經證實ATM 與心血管疾病之間的關系, 但是仍需要更多的后續研究進行驗證。
關節炎與ATM 蛋白有相關性的研究,2009 年研究發現類風濕性關節炎患者都缺少DNA 修復酶ATM, 分別探索ATM及其相關信號通路都被抑制;同時他進行了ATM 蛋白的過表達實驗通過激活DNA 修復通路可有效治療關節炎[80]。 另外兒童誘發性關節炎常常發生在16 歲之前,該病極大可能發展成慢性風濕病甚至導致殘疾,而近期報道ATM 蛋白與兒童誘發性關節炎高度相關[81]。 20 世紀初,ATM 缺失小鼠也被用于作為骨質疏松模型使用, 研究顯示ATM 缺失導致骨骼質量下降、骨骼形成減緩和成骨細胞分化/成熟失敗等表型[82]。
3 與衰老調節相關的信號通路分析
衰老是骨骼肌衰減癥的發病機制的基礎, 與肌肉退行性進程性的改變相關[8],本節主要討論可能影響衰老的具體機制,包括胰島素類生長因子信號通路、雷帕霉素受體信號通路、線粒體和氧化應激反應、細胞衰老、蛋白質穩態、生物節律等方面[83],比對ATM 蛋白與其相關作用。
3.1 ATM 與胰島素
大部分AT 病人血清指標中胰島素樣生長因子-1(Insulin-like growth factor-1,IGF-1)含量偏低,推測是由IGF-1及其相關蛋白影響機體生長和體重增長所造成的[45,84]。 Schubert[45]認為9/16 的患者IGF-1 含量低于同齡對照的前3%,而且13/16 的患者IGF-1 主要結合蛋白IGFBP-3 濃度顯著較低。 Voss[85]測定了24 名患者的IGF-1 水平,結果顯示其中10位患者的濃度(41.7%)低于同齡前3%,Kieslich[84]的研究也表明6 名AT 病人中IGF-1 的含量低于前3%。Pommerening[14]測定了激素相關指標,發現IGF-1、皮質醇和脫氫表雄酮顯著低于對照組。 與此同時,提取AT 病人成纖維細胞并進行離體培養,發現IGF-1-sCLU(secretory clusterin)蛋白水平顯著低于對照組,可能是IGF-1 含量偏低所引起的[86]。
3.2 ATM 與mTOR 通路
3.2.1 與mTOR 上游通路
Akt 是mTOR 通路中的一個核心因子, 研究發現Akt 和ATM 基因敲除模型小鼠具有許多相似表型, 如: 生長停滯、不育癥、免疫系統缺陷和胰島素抵抗[87-88]。 通常情況下,ATM蛋白含量與細胞中Akt 磷酸化水平無相關性。 當機體受到胰島素/IGF-1 等條件刺激時ATM 缺失會直接影響Akt 的磷酸化[25,89-90]。 Ching[90]對比目魚肌細胞和C2C12 肌管采取10 nM的IGF-1 刺激,時長為20 min,結果顯示ATMKD 的C2C12 肌管顯著下調Akt 的S473/T308 磷酸化水平;與ATM+/+小鼠的比目魚肌細胞相比,ATM+/-的AktS473/T308 磷酸化水平發生明顯的阻礙作用, 進一步檢測發現p-mTOR 蛋白含量也顯著下調,而總體mTOR 沒有改變。
為了探尋ATM 與IGF-1R 的直接聯系, 研究者將完整ATM 全片段的cDNA 轉染到AT 突變細胞中[91],通過實驗技術Westernblot 驗證8 個細胞GM5849 的cDNA 全部成功轉染,證實有全面表達的ATMcDNA 可增加IGF-1R 的表達水平[92]。 氨基末端激酶(Jun N-terminal kinase,JNK)誘導IRS-1 S307 磷酸化,阻斷胰島素信號通路Ching[90]利用激酶反應試劑盒檢測野生型和雜合型中比目魚肌的PI3K 活性,結果顯示IGF-1 刺激下的PI3K 活性在ATM+/-中偏低(p=0.053)。 Viniegra[93]研究ATM 對Cos 細胞胰島素刺激的Akt S473 的磷酸化有直接作用, 進一步探究是否通過抑制PI3K 能夠阻礙Akt 的磷酸化,與預期結果相一致,PI3K 的各種抑制劑(200 nM 渥曼青霉素、2 μML Y294002、2 nM 咖啡因) 的加入完全阻礙S473 的磷酸化,即抑制ATM-PI3K-Akt 通路中的PI3K 可直接阻礙Akt 的磷酸化進行。
3.2.2 ATM 與mTOR 下游通路
在活體雷帕霉素影響的研究中編碼翻譯過程受到許多磷酸化所調控, 如核糖體S6 蛋白和其激酶p70S6K1 以及eIF-4F 結合蛋白4E-BP1。 Ching 進行3 項單獨實驗驗證ATM 在IGF-1 刺激下促使S6K1 磷酸化下調:1)IGF-1 刺激的S6K1磷酸化位點在T389,C2C12 細胞ATM shRNA 中S6K T389 磷酸化水平顯著下降;2) 正常的C2C12 細胞 (ATM+/+) 培育在KU55933 中,通過阻礙胰島素通路中的PI3K,進而抑制通路進程來降低S6K1 的磷酸化水平;3)在觀察只有一半功能的雜合型ATM+/-比目魚肌細胞,在IGF-1 刺激下S6K1 磷酸化水平顯著低于野生型對照[90]。 Burnett[94]報告ATM 類似家族的相關激酶有助于促進mTOR 下游S6K1 和4E-BP1 磷酸化。Yang[95]在研究胰島素刺激下蛋白質合成通路過程中,發現誘導eIF4E的結合蛋白4EBP1,ATM 能在4EBP1 的S111 位點發生磷酸化,當ATM 缺失的離體細胞,顯著下調4EBP1 從eIF4E 分離水平。 與此結果相一致的研究Kuang[96]在ATM-/-小鼠中發現ATM 的功能缺失下調mTOR 信號通路的4EBP1 水平。
基于以上所述,基本構建出ATM 阻礙/抑制骨骼肌蛋白質合成的可能路徑(見圖3)。 ATM 缺失/下調可能發生的假設路徑:下調ATM→下調IGF-1→下調IGF-1R→下調IRS-1(通過下調Y612 或上調S307)→下調PI3K→下調Akt(通過下調S473 和T308)→下調mTOR→下調S6K1 和下調4E-BP1 的分離率→骨骼肌蛋白質合成[69]。
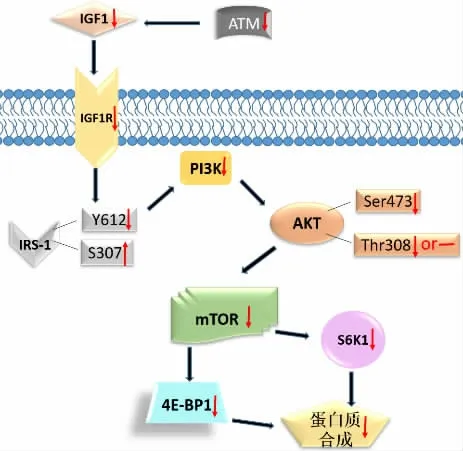
圖3 ATM 阻礙骨骼肌蛋白質合成的路徑假設[69]
3.3 ATM 與ROS
ATM 蛋白在調控ROS 中發揮突出作用,AT 的嚙齒動物和細胞模型/系都有氧化應激水平升高的共同特性,ATM 蛋白缺失導致的DNA 雙鏈斷裂和機體ROS 急劇升高,是引起AT病人和動物模型、細胞模型衰老的主要原因[4,97]。
早期研究表明突變的ATM 缺乏動物實驗中提升機體的氧化大分子及ROS 水平:ATM-/-小鼠與正常小鼠大腦區域和特定神經元的活性氧進行對比發現,ATM-/-小鼠的浦肯野細胞和黑質多巴胺能神經元的超氧化物水平顯著升高[98];另一組實驗發現ATM-/-腦中含硫醇化合物的水平發生了變化, 同時ATM-/-小腦中的硫氧還蛋白、過氧化氫酶和錳超氧化物歧化酶活性也發生了顯著升高現象[99];4 月齡的ATM-/-小腦呼吸率和造成NADPT 和NADP+氧化還原總體水平的降低等[100]。 近期研究也證實了ROS 升高現象,Weyemi[101]等首次證明NADPH氧化酶4(NOX4)參與調節AT 疾病,并報道AT 細胞中有較高水平的NOX4,ATM 的缺失直接導致NOX4 的高表達與正常細胞進行比較;將ATM 突變小鼠中分離出的小腦星形膠質細胞,抗氧化劑GSH 和谷胱甘肽-s-轉移酶的表達均下降[102-103]。
有研究認為ATM 缺陷小鼠的神經細胞、 培養的細胞系等,可通過抗氧化劑的處理改善和延緩由ROS 引起的衰老表型。 當添加異吲哚胺硝基可以防止ATM 缺陷小鼠的浦肯野細胞死亡,并將樹突發育水平提高到野生型水平[104];當注射fulvene-5 后,降低癌癥風險,并治療神經退行性的癥狀[101];另外添加n -乙酰- l-半胱氨酸(NAC) 可恢復ATM-/-星形膠質細胞的正常增殖(ATM 是星型交織細胞生長所必須的,當通過調控ROS 水平恢復ATM 功能),恢復AT 突變星形膠質細胞中的谷胱甘肽水平[102-103]。
3.4 ATM 與DNA 損傷修復
多種代謝活動都會引起哺乳動物細胞中DNA 雙鏈的斷裂,導致突變和細胞轉化失調,ATM 缺失直接導致機體、組織和細胞中DNA 雙鏈的斷裂, 除了受到持續激增的ROS 影響外,還受到修復能力下降和速率減緩的限制,主要的DNA 損傷應激信號成分是蛋白激酶ATM 和ATR[105-106]。 Song[107]等通過AT 病人觀察發現其先天免疫系統的膠質細胞受到影響,ATM 活性的喪失導致DNA 修復的減慢及基因組DNA 細胞質片段的累積增多。 研究認為衰老和老齡化會導致以ATM 為主導因素的DNA 損傷應激過程加劇, 通過激活ATM 可緩解相關癥狀和進程[106]。 因此,ATM 可直接或間接調控相關信號通路,進行修復DNA 損傷后的修復。
1)ATM 參與調控核區泛素化作用促進DNA 損傷修復。本世紀初,多個獨立研究團隊都進行了相關研究,證實DNA 雙鏈斷裂后會自發激活ATM,ATM 活化后會將H2AX 上蘇氨酸S139 的位點磷酸化,生成γH2AX,最終進行DNA 的損傷修復[108-110]。2)ATM 調控其他染色質修飾實現修復功能。①H2BUb 下調直接影響細胞中非同源性末端連接(Nonhomologous End Joining,NHEJ) 和同源重組 (Homologous Recombination,HR)修復[111]。 H2B 被發現在DNA 雙鏈斷裂附件發生泛素化,并且受到RNF20-RNF40 異二聚體的催化。②ATM 激活KAP1(KRAB-associated protein 1, also known as TIF1beta, TRIM28 or KRIP-1)的磷酸化釋放染色質進行修復[112]。KAPI 是一種轉錄輔抑制因子,它與組蛋白甲基轉移酶和組蛋白去乙酰化酶復合物協同作用,共同促進染色質的致密化。 KAP1 磷酸化依賴于ATM,干預KAPI 和CHD3 核小體的重構進而促進染色質的釋放[113]。 3)ATM 被DNA 單鏈斷裂(single-strand breaks,SSBs)修復DNA 雙鏈斷裂。DNA SSB 既是DNA 自發失穩狀態下的結果,也是DNA 修復過程中的中間產物,所以SSB 的修復必然發生在DNA 復制之前。 CHK1 和CHK2 是ATM/ATR 主要的反應靶點,與ATM 和ATR 一起,通過各種基質作用于減少循環依賴激酶CDK[114]。 研究報道ATM 可被SSB 所激活同時延長G1 細胞周期延遲,從而更多的時間進行DNA 損傷修復[115]。
3.5 ATM 與小腦神經退行性
AT 是一種復雜的中樞神經退行性病變的疾病,AT 病人的神經變化與ATM 激酶的缺失或部分缺失有密切關聯,這一過程可轉化為小腦神經退行性變和AT 神經肌肉失用相關,最終導致肌肉刺激不足或錯誤,進而促使肌肉退化[116]。 ATM 蛋白缺失與機體神經退行性變化直接相關, 因而AT 病人與AT動物模型都存在肢體控制能力下降, 不能行走或行走緩慢等表征[4,26]。Ruiz-botero[12]等報道AT 病人出現運動失調,四肢末梢開始抖動等;早期文獻也報道AT 病人手臂出現顫抖而且精準度下降,行走困難等[16];這些現象都顯示神經對肌肉控制能力下降,提示ATM 蛋白缺失對小腦以及神經網絡有損傷。
4 ATM 嚙齒動物模型
目前關于衰老的動物模型構建: ①自發性包括自然衰老和快速老化模型(SAMP8 應用最廣);②誘導性模型包括D-半乳糖致亞急性衰老模型、β 淀粉樣蛋白誘導模型、去胸腺誘導模型、γ 射線誘導模型、氯化鋁誘導和亞硝酸鈉等藥物誘導模型;③轉基因模型例如應用CRISPR/Cas9 等[117]。 本節主要介紹ATM 蛋白缺失的嚙齒動物模型,通過目的基因的敲除抑制ATM 蛋白表達的統計, 為之后衰老及骨骼肌衰減的研究作模型參考。

表2 ATM 嚙齒動物模型構建庫
為了探索ATM 蛋白缺失對機體的整體影響,多種ATM-/-的嚙齒動物模型相繼敲除成功[4]:大鼠敲除ATM-/-第13 外顯子模型無ATM 蛋白表達; 小鼠模型除了ATM△SRI/△SRI7636del9(129SvJ C57BL/6J)表達少量蛋白以外,其余模型均無ATM 蛋白表達。 有部分實驗認為ATM 缺失不會造成共濟失調和小腦神經退行變化[28],然而大部分均認為有顯著作用。ATM 缺失造成運動能力下降 (旋轉臺和開放區域實驗能力下降)[28,118];小腦結構發育過程中被破壞[29]、小腦萎縮[26];神經元及網絡結構惡化[26,29,104]和突觸傳遞退化(多巴胺黑紋狀體嚴重退化和末端紋狀體不對稱[119]、神經遞質發生退化[26];Purkinje 細胞的異常遷徙和增多[27]、樹突狀分支減少和不規則現象[26,29,104]和胞內溶酶體數量增加[99]等;小腦組織和細胞中氧化應激標志物增加[99,120]和線粒體功能下降[26-27]。

圖4 ATM-/-小鼠創建的路徑研究
5 小結
衰老與抗衰老是人類目前科學研究重要命題, 本文通過ATM 蛋白缺失(人、動物模型、細胞)與骨骼肌衰減癥的表征、衰老相關疾病、引起骨骼肌衰減癥的內在機制探索,初步得出以下結論:1)AT 病人(ATM 動物模型)與骨骼肌衰減癥具有一致的表型(骨骼肌質量和肌力下降、運動能力減弱),并且運動訓練可扭轉2 種疾病的病癥。 2)ATM 蛋白缺失與衰老引起的老年癡呆、糖尿病、癌癥、骨質疏松等老年病具有高度相關性,ATM 參與和保持骨骼肌功能(胰島素、mTOR、氧化應激、DNA損傷修復、神經退行性)與衰老引起的功能下降機制具有一致性。 3)梳理現有的ATM 動物模型發現骨骼肌質量下降、運動能力逐步消失、小腦功能失調和退行性的改變,因此建議ATM蛋白缺失模型可作為骨骼肌衰減癥的動物模型。
猜你喜歡
興趣閱讀·興趣作文與閱讀(低年級)(2025年8期)2025-08-18 00:00:00
童話王國·奇妙邏輯推理(2024年5期)2024-06-19 16:03:38
體育科技文獻通報(2022年3期)2022-05-23 13:46:54
遼金歷史與考古(2021年0期)2021-07-29 01:06:54
中學生數理化·七年級數學人教版(2020年10期)2020-11-26 08:24:50
學苑創造·A版(2020年9期)2020-10-13 09:41:02
數學物理學報(2020年2期)2020-06-02 11:29:24
科技傳播(2019年22期)2020-01-14 03:06:54
民用飛機設計與研究(2019年4期)2019-05-21 07:21:24
光學精密工程(2016年6期)2016-11-07 09:07:19
