配體引導銅基配合物的合成、表征及催化性能
2023-07-14 01:21:08于衛東
人工晶體學報 2023年6期
張 銀,于衛東
(1.長沙師范學院初等教育學院,長沙 410000;2.湖南工商大學理學院,長沙 410000)
0 引 言
近年來,配位聚合物的應用不斷拓展,受到了廣泛關注[1-2]。根據結構特點的不同,配位聚合物可應用于氣體或污染物的吸附和分離[3]、光學[4]、磁性[5]、傳感[6]與電化學[7]等領域。而隨著綠色催化概念的提出,通過對配體的設計而將配位聚合物應用于催化領域,則成為了最為廣泛的應用[8]。雖然,金屬中心、合成溫度和時間、反應體系的pH值等因素均會對配位聚合物的結構造成影響,但是配體的設計及選取可以直接改變配位聚合物的結構特性和性能,是此類材料合成的關鍵技術[9-10]。酰胺類化合物由于其易制備且結構中往往含有多個配位位點而被選作配體[11]。配位原子在結構中的不同位置可以誘導形成不同的配合物結構[12]。比如形成單分子化合物,或是金屬之間再通過橋連氧連接形成金屬簇合物[13-14]。
吡啶基團由于其良好的配位能力而常常被用作配體的修飾基團[15-16]。將吡啶與酰胺結合形成的配體,因為吡啶的引入而增添了配位位點,從而使其具有配位多樣性[17]。渤海大學的王秀麗教授課題組在此方面做了大量工作,其中基于半剛性的吡啶酰胺配體的配合物展現出了較強的熒光傳感能力和電化學傳感能力[18-19],而基于柔性的吡啶酰胺配體的配合物則展現出了較高的熒光猝滅性能[20]。
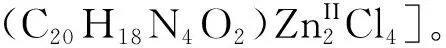
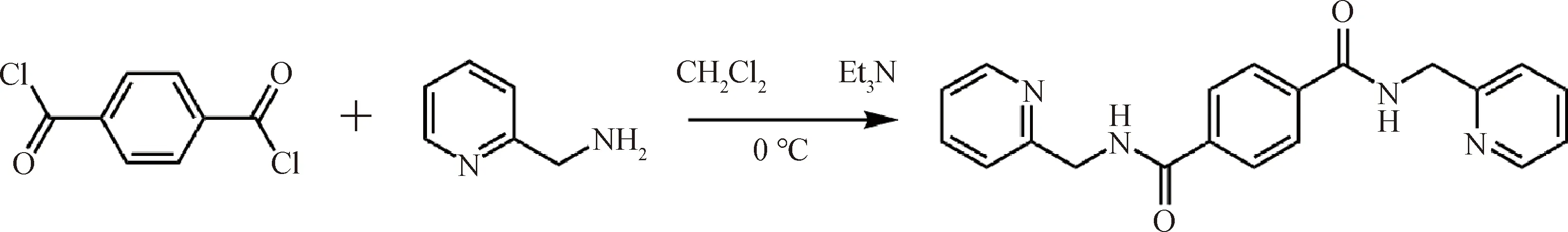
圖1 配體L1的合成路線Fig.1 Synthetic route of ligand L1
1 實 驗
1.1 實驗原料
乙醇、乙腈、甲醇、甲苯、氯仿、四氫呋喃、NN-二甲基甲酰胺、二甲基亞砜、丙酮、二氯甲烷、三乙胺均為分析純,購自國藥集團;氯化銅(純度98%)、氯化鋅(純度98%)、醋酸銅(純度98%)、對苯二甲酰氯(純度99%)、2-(氨甲基)吡啶(純度99%)、1-(2-吡啶偶氮)-2-萘酚(純度99%)、六氟磷酸銨(純度99%)購自安耐吉化學。
1.2 儀器和測試方法
1H NMR譜圖在Bruker Advance 400 MHz 測試儀上測試得到,氘代試劑加入了四甲基硅烷為內標;質譜數據在以電噴為離子源的Bruker Compact四級桿飛行時間質譜上測試得到,數據分析在Bruker Isotope Pattern軟件中模擬;晶體結構數據和精修數據如表1所示,單晶X射線衍射數據在Bruker APEX-II衍射儀上測試得到。
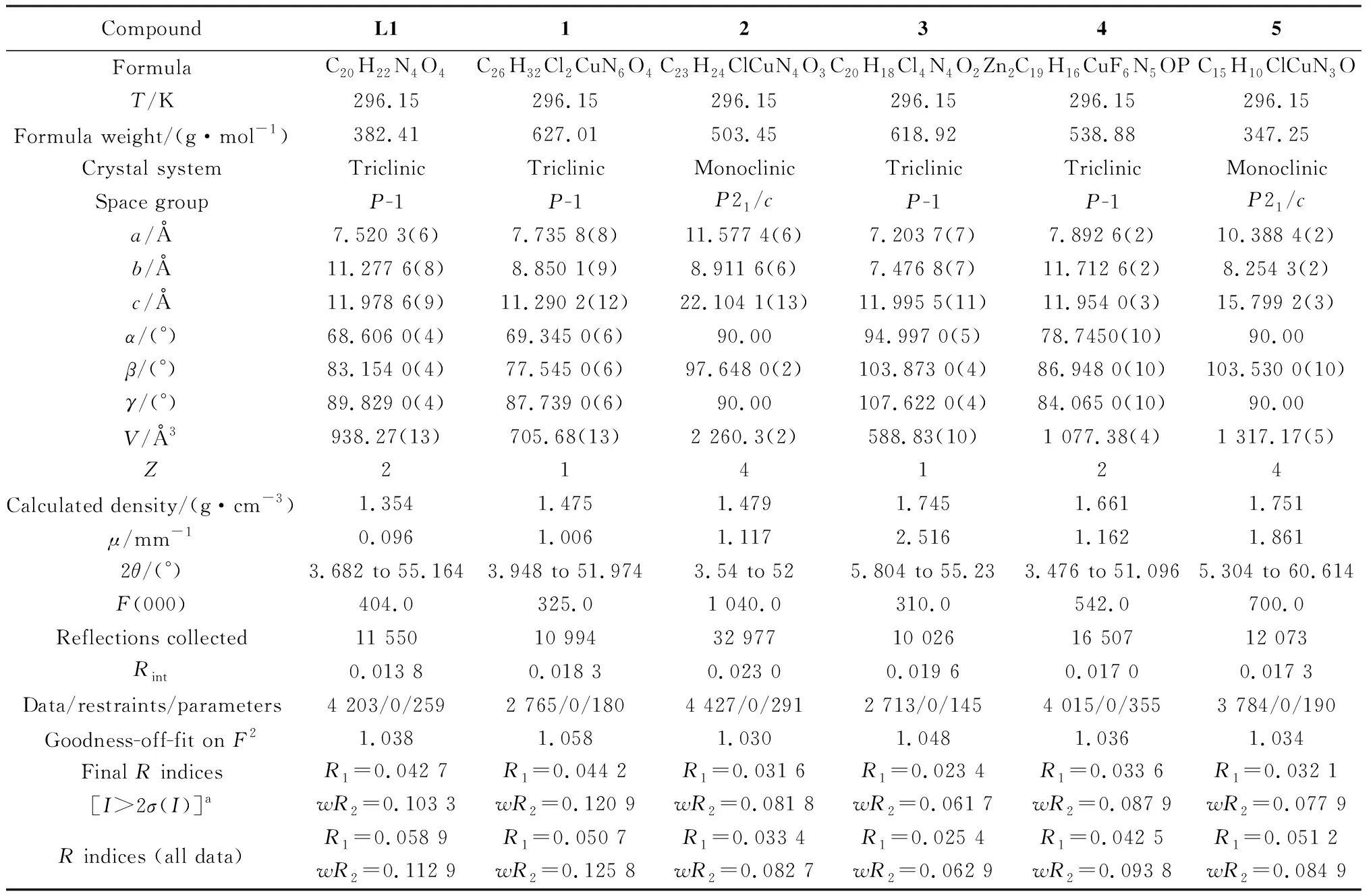
表1 配體L1以及配合物1~5的晶體數據和結構精修數據Table 1 Crystals data and structure refinement parameters for ligand L1 and complex 1~5
1.3 實驗步驟
配體L1的合成:配體L1根據文獻[21]并對其實驗步驟稍加優化后進行。將含有對苯二甲酰氯(4 mmol)的二氯甲烷(20 mL)溶液逐滴加入到含有2-(氨甲基)吡啶(8.8 mmL)和三乙胺(8.8 mmol)的二氯甲烷(40 mL)溶液中。混合溶液在冰水浴中反應3 h,并產生了大量的白色沉淀。將沉淀溶于甲醇中重結晶,得到配體L1。產率約為85%。將配體L1溶于甲醇中,緩慢揮發幾天后得到無色針狀晶體用于單晶X射線衍射測試。
配合物1的合成:將含有氯化銅(4.4 mmol)的甲醇溶液(30 mL)逐滴滴入含有配體L1(4.4 mmol)的甲醇溶液(20 mL)中。將混合溶液在室溫下攪拌5 h,得到綠色沉淀。過濾并用大量的甲醇沖洗。將綠色沉淀溶于DMF中,并用不良性溶劑丙酮進行氣相擴散結晶,幾周后得到深藍色塊狀晶體1,產率大約為45%。
配合物2的合成:合成步驟同配合物1。只是結晶時將配合物溶于DMSO中,并用不良性溶劑丙酮進行氣相擴散結晶,幾周后得到深藍色塊狀晶體2,產率大約為25%。
配合物3的合成:合成步驟同配合物1,只是將氯化銅換成了氯化鋅。結晶過程選用良性溶劑DMF或DMSO均可,再用不良性溶劑丙酮液相擴散,幾周后得到大量的無色塊狀晶體,產率大約為60%。
配合物4的合成:將醋酸銅(4.2 mmol)、配體L2(4 mmol)和六氟磷酸銨(5 mmol)溶于乙腈溶液(50 mL)中,回流攪拌5 h。然后將反應后的溶液過濾,濾液緩慢揮發,7 d得到大量的長棒狀黑色晶體,產率約為80%。
配合物5的合成:合成步驟同配合物4,只是將醋酸銅和六福磷酸銨替換為將氯化銅(4.2 mmol),揮發結晶約7 d后得到大量的長棒狀黑色晶體,產率約為85%。
2 結果與討論
2.1 配合物1~5的晶體結構描述
利用單晶X射線衍射對配合物的結構進行了比較分析,相關單晶數據列于表1中。圖2則展示了五種配合物的晶胞以及中心金屬的配位環境。1屬于三斜晶系P-1空間群,晶胞參數為a=7.735 8(8) ?,b=8.850 1(9) ?,c=11.290 2(12) ?,α=69.345 0(6)°,β=77.545 0(6)°,γ=87.739 0(6)°,V=705.68(13) ?3。根據1的分子式以及BVS價鍵理論[22]計算,可以得到金屬中心Cu為+2價。金屬中心Cu2+呈現出了六配位模式,一個Cu2+分別與兩個L1中吡啶環上的N[Cu1—N1=Cu1—N1A=2.048(2) nm]和羰基O[Cu1—O1=Cu1—O1A=2.565(2) nm]以及兩個Cl-配位[Cu1—Cl1=Cu1—Cl1A=2.297 1(7) nm]。2屬于單斜晶系,P21/c空間群,晶胞參數為a=11.577 4(6) ?,b=8.911 6(6) ?,c=22.104 1(13) ?,α=γ=90.00°,β=97.648(2)°,V=2 260.3(2) ?3。根據2的分子式以及BVS價鍵理論計算,可以得到金屬中心Cu為+1價。金屬中心Cu2+呈現出了四配位模式,Cu+與兩個L1中吡啶環上的N[Cu1—N1=2.078 6(17) nm;Cu1—N2=2.054 1(5) nm]以及兩個Cl-配位[Cu1—Cl1=2.428 7(5) nm;Cu1—Cl1A=2.373 9(5) nm]。其中Cl-分別與兩個Cu+相連,具有橋連作用。由于1和2的合成步驟相同,只是結晶時選用的良性溶劑不同。因此推測造成兩種配合物結構差異的原因可能是2在結晶過程中DMSO的還原性和Cu2+的弱氧化性發生了反應,使得Cu從+2價還原成了+1價,并導致其分子在空間中的排列方式也發生了變化。1中的每個L1配體與Cu2+交替排列,形成了“之”字型的扭曲一維鏈狀結構。2則呈現出了配體平行排列,而相鄰的配體之間由Cu+和Cl-橋連相接,形成了二維的網狀結構。
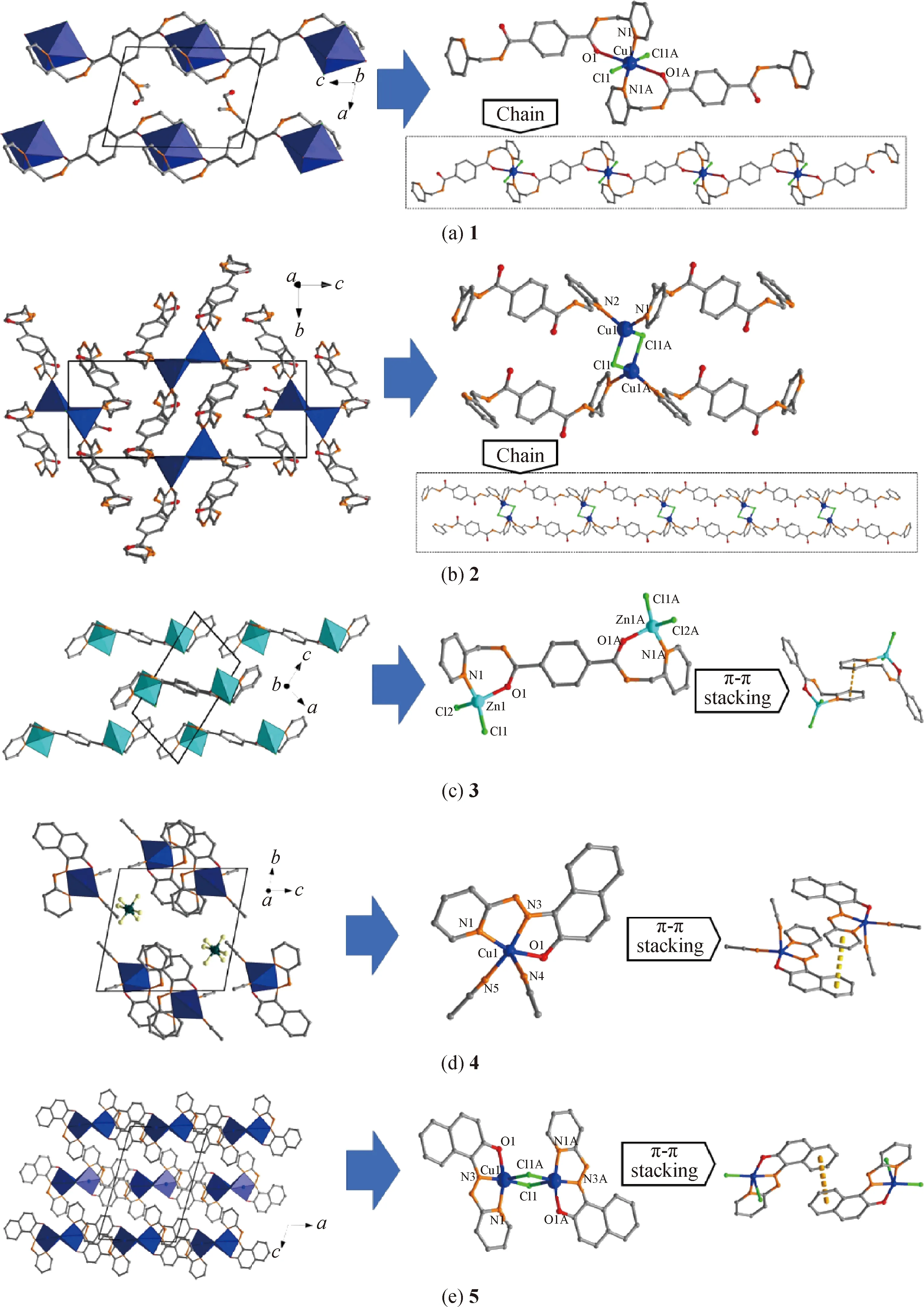
圖2 配合物1~5的晶體結構示意圖Fig.2 Crystal structure diagrams of complex 1~5
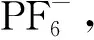
為了比較L1在不同配合物晶體中的不同,將配體L1的吡啶環旋轉方向進行比較(見圖3)。從圖中可以看出,配體L1單獨結晶時與形成配合物時吡啶環的旋轉方向發生了明顯的改變,這說明在與金屬發生配位反應時,由于金屬的配位作用,配體發生了旋轉。而對比不同配合物的L1配體可以發現,當中心金屬的配位模式不同時,吡啶環的旋轉也會不同。
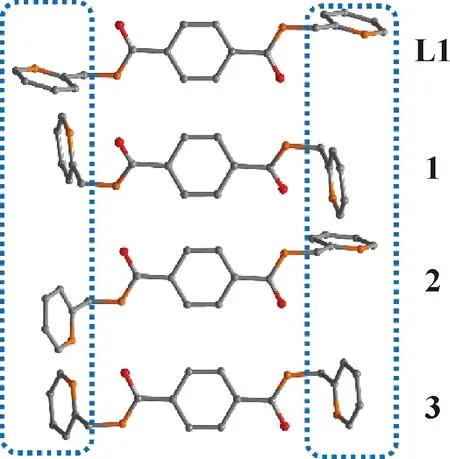
圖3 配體L1在形成不同配合物時吡啶環的翻轉示意圖Fig.3 Schematic inversion of the pyridine rings of L1 in different complexes
2.2 核磁表征
由于Cu2+對核磁信號的影響作用,本文僅僅討論了配體L1和配合物3的核磁峰位移變化。從圖4中可以看出,當配體與Zn2+配位后配體上H的出峰發生了不同程度的位移變化。吡啶環上除鄰位H(a處)外,其余H信號均向低場區移動。亞甲基上的H(e處)由于Zn2+配位后的金屬屏蔽作用,其信號也向低場區發生移動。值得一提的是,雖然L1和3的核磁均在d-DMSO中獲得,但是氨基上的H的信號僅在3的核磁氫譜中顯示。這說明在L1中,氨基中的H具有較高的活性,因此沒有在核磁氫譜中顯示,但是當與Zn2+配位后,配位鍵的存在,使氨基中的H降低了活性,從而在核磁氫譜中得以顯示。
圖4 配合物3(上)和配體L1(下)在d-DMSO中的1H NMR圖譜Fig.4 1H NMR spectra of complex 3 (up) and ligand L1 (down) in d-DMSO
2.3 質譜表征
除核磁外,電噴霧質譜也常被用來對配合物進行表征[23-24]。對1和2進行了電噴霧質譜分析,從圖5(a)中可以看出,配體L1的在正離子模式下出現了兩個主峰,分別對應的離子碎片為[L1+H]+和[L1+Na]+。而1在正離子模式下出現了兩個主峰,分別對應的離子碎片為[L1+Cu+Cl]+和[L1+Cu2+Cl3]+。從圖中可以看出,電噴霧質譜測試過程中的高電壓等極端條件導致1的鏈狀結構遭到破壞,使測出的離子碎片分別為一個配體配位一個或兩個Cu2+。而不同于1,2的質譜圖中主要得到了3個主峰,分別對應離子碎片為[L1+Cu]+、[L1+Cu+Cl]+和[L1+Cu+DMF]+。由于2中的Cu是+1價,所以在質譜測試過程中,Cu離子仍然保持了+1的價態,而離子碎片[L1+Cu+Cl]+則可能是由于測試過程中Cu+氧化而變成了+2價。通過質譜分析可以看出,雖然1和2所使用的配體都相同,但是由于銅離子價態不同,使得其測試結果也展現出了不同。
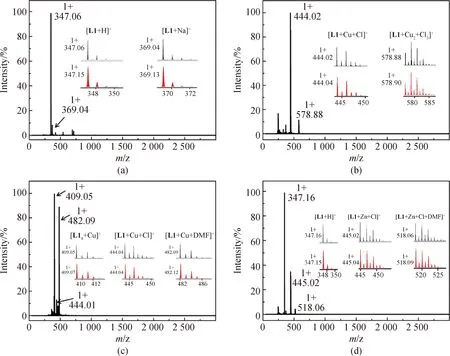
圖5 配體L1(a)、配合物1(b)、配合物2(c)和配合物3(d)在DMF和甲醇混合溶液中的電噴霧質譜圖(正離子模式)Fig.5 ESI-MS spectra of ligand L1 (a), complex 1 (b), complex 2 (c) and complex 3 (d) in DMF and CH3OH (positive mode)
2.3 催化性能分析
為了探討配合物不同的結構與性能的關系,選取了硫醚氧化生成亞砜和砜的催化體系作為研究對象。首先對催化條件進行篩選(以配合物3作為催化劑),如表2所示。當用O2或者是TBHP(叔丁基過氧化氫)作氧化劑時,催化轉化率較低,但是當選用雙氧水作為氧化劑時,催化反應卻展現出了較好的催化轉化率。這主要是由于氧氣的氧化性弱于雙氧水,從而較難氧化底物。TBHP和H2O2相比,分子體積較大,而空間位阻影響了其與金屬結合,使得其氧化性較弱。此外,催化反應的溫度也對此反應有影響作用,溫度越高在同等條件的催化轉化率越高。不同的反應溶劑對反應也具有不同的影響,在乙腈和乙醇中的反應具有較高的催化轉化率,而當使用氯仿、四氫呋喃和甲苯當溶劑時,催化轉化率較低。因此,催化反應選用H2O2作為氧化劑,乙醇作為反應溶劑,在60 ℃條件下進行。
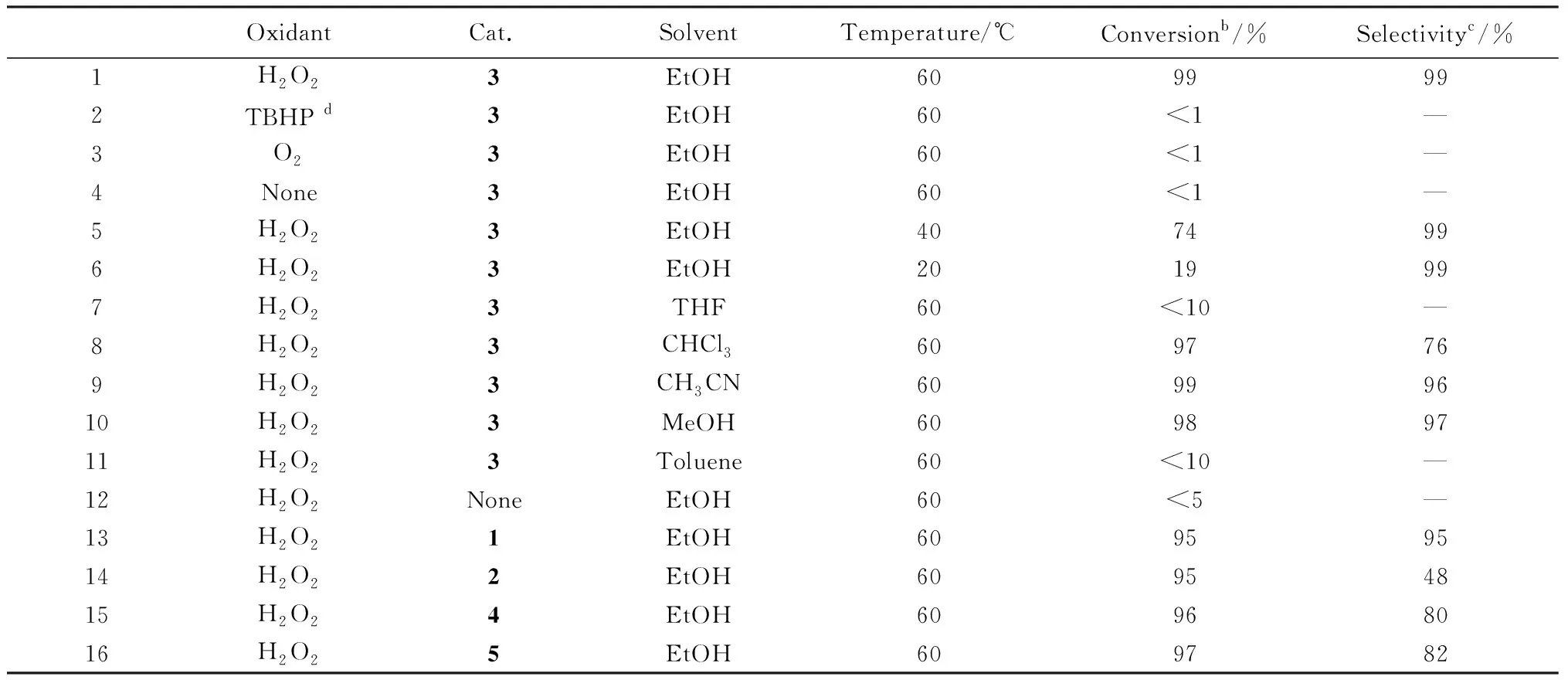
表2 不同催化條件下的硫醚氧化成亞砜和砜的催化反應aFig.2 Catalytic reactions of thioether oxidation to sulfoxide or sulfone under different conditionsa
對比幾種不同配合物在相同的催化條件下催化產率可以看出,配合物3具有最高的催化效果。這可能和不同金屬中心的催化活性有關系。與文獻[25-27]報道類似,對于硫醚的氧化催化反應,Zn比Cu具有更高的催化活性,因此配合物3比其他配合物的催化轉化率高。對比幾種中心金屬為Cu的配合物可以發現配合物1、4和5的催化效率都高于配合物2。從結構中分析可知,配合物2結構中的Cu為+1價態,而其余配合物中Cu的價態均為+2價,因此金屬中心的不同價態對配合物的催化效果也具有明顯的影響。而配合物1、4與5比較發現,配合物1的催化效果優于其他兩個,造成這種催化差異的主要原因可能是分子的堆積方式不同。配合物4和5的配體是單齒配體,所以并沒有形成多維結構,而因為配體中存在大π鍵,配體之間存在π鍵的堆積作用。這使得分子之間的空隙較小,底物和氧化劑較難與金屬離子中心結合,降低催化活性。基于以上實驗結果和以前的文獻,推測出了一個可能的催化機理,其中催化劑以配合物3為例,如圖6所示。首先,配合物3與雙氧水結合形成過氧配合物,并且具有更高的氧化活性。過氧配合物與甲基苯基硫醚結合后脫去水形成了中間產物甲基苯基亞砜。甲基苯基亞砜再與過氧配合物結合,脫去水形成了最終產物苯甲砜。
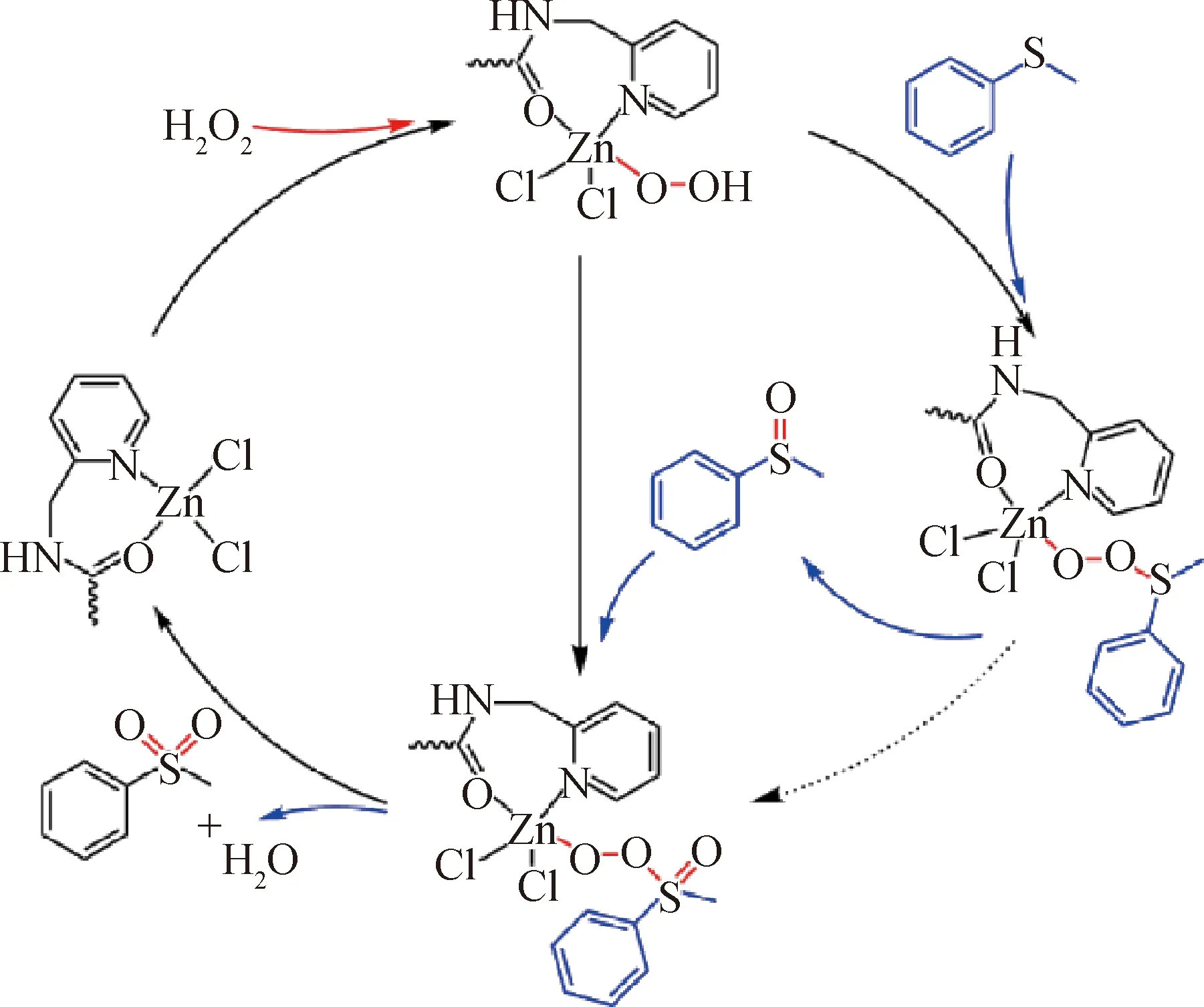
圖6 可能的反應機理Fig.6 Proposed reaction mechanism
3 結 論
1)選用酰胺類配體與Cu反應,通過改變結晶條件,發現得到的配合物1和2結構中的Cu離子價態不同,同時配位模式和空間構型也發生了變化。但是當換成同樣具有多配位模式的Zn2+,或是換成偶氮類配體進行反應時,卻只能得到一種構型的配合物。
2)通過質譜分析也同樣發現,1的離子碎片峰僅含有+2價的Cu離子碎片峰。而2的離子碎片峰中則含有+1價的Cu離子碎片峰。
3)利用5種配合物催化氧化苯甲基硫醚,結果發現3具有最好的催化效果,60 ℃下反應1.75 h即可將苯甲基硫醚完全催化氧化為砜。而1和2相比,1則具有更好的催化活性。
4)基于催化結果,推測了一個可能的催化反應機理,即氧化劑先與配合物的金屬中心進行結合形成過氧配合物,然后再氧化硫醚生成砜。
