氣相色譜-串聯質譜法同時測定當歸中35種禁用農藥殘留量
2023-07-04 16:23:32吳福祥李堅朱仁愿閆君陳婷丁輝張文
安徽農業科學 2023年5期
關鍵詞:氣相色譜
吳福祥 李堅 朱仁愿 閆君 陳婷 丁輝 張文
摘要 ?[目的]利用氣相色譜-串聯質譜(GC-MS/MS)建立同時測定當歸中35種禁用農藥殘留量的分析方法。[方法]色譜柱為HP-5MSUI(30 m×0.25 mm,0.25 μm);載氣流量1.2 mL/min;進樣量1 μL,不分流進樣;進樣口溫度280 ℃;程序升溫。樣品以乙腈直接提取,采用GC-MS/MS的多反應監測分析模式,通過基質匹配標準溶液降低基質干擾,內標法定量。[結果]35種禁用農藥在各自濃度范圍內線性良好,相關系數均不低于0.985 2,檢出限為0.155 5~7.173 9 μg/kg,定量限為0.513 2~23.673 9 μg/kg,精密度RSD均小于14%(n=6)。平均加樣回收率為83.86%~122.80%,RSD為3.8%~24.6%(n=5)。[結論]該方法快捷、準確,可用于當歸中禁用農藥殘留的定性和定量分析。
關鍵詞 ?當歸;禁用農藥殘留量;氣相色譜-串聯質譜法
中圖分類號 ?S 481+.8 ??文獻標識碼 ?A ??文章編號 ?0517-6611(2023)05-0198-05
doi: 10.3969/j.issn.0517-6611.2023.05.045
開放科學(資源服務)標識碼(OSID):
Simultaneous Determination of 35 Banned Pesticide Residues in Angelica sinensis by GC-MS/MS
WU Fu-xiang, LI Jian, ZHU Ren-yuan et al
(Lanzhou Institute for Food and Drug Control/Key Laboratory of Pesticide and Veterinary Drug Monitoring for State Market Regulation /Gansu Engineering Research Center for Monitoring Exogenous Harmful Residues in Traditional Chinese Medicines, Lanzhou,Gansu 730050)
Abstract ?[Objective] A method for simultaneous determination of 35 banned pesticide residues in Angelica sinensis was established by gas chromatography-tandem mass spectrometry (GC-MS/MS). [Method] Chromatographic column was HP-5MSUI(30 m×0.25 mm,0.25 μm);gas traffic was 1.2 mL/min;injection volume was 1 μL, no split injection;injection temperature was 280 ℃;the program heats up.The samples were extracted directly with acetonitrile, and the multi-reaction monitoring and analysis mode of GC-MS/MS was used. Matrix interference was reduced by matrix matching standard solution, and the internal standard method was used for quantification. [Result] The 35 banned pesticides had good linearity within their respective concentration range, the correlation coefficient was not less than 0.985 2,the limit of detection was 0.155 5-7.173 9 μg/kg, the limit of quantification was 0.513 2-23.673 9 μg/kg, the RSD of precisions were all less than 14%(n=6), the average recovery rate was 83.86%-122.80%, and the RSDs were 3.8%-24.6%(n=5). [Conclusion] The method is rapid and accurate, and can be used for qualitative and quantitative analysis of banned pesticide residues in Angelica sinensis.
Key words ?Angelica sinensis;Banned pesticide residues;Gas chromatography tandem mass spectrometry (GC-MS/MS)
中醫藥是我國文明的寶貴遺產,在幾千年的歷史中不斷進步,為人類的健康作出了重大的貢獻。中藥作為中醫藥體系的核心組成部分,一直以來都受到人們的充分重視。甘肅省是我國中藥材的主產區之一,素有“千年藥鄉”和“天然藥庫”的美譽,中藥資源十分豐富。據2019年統計,甘肅省中藥材種植面積31.33萬hm2,產量52萬t,產值超32億元;其中,當歸、板藍根、黨參等道地藥材產量分別占全國市場的95%、65%、60%,種植面積及產量居全國前列[1]。近年來,中藥的質量安全問題阻礙了我國中藥產品進入國際市場,主要原因就是農藥殘留、重金屬及真菌毒素等外源性污染問題。其中,農藥殘留污染主要有3個方面:一是種植過程中,施用農藥防治病蟲害,造成農藥濫用的直接污染;二是土壤和空氣中殘留的農藥造成的間接污染;三是采收加工、儲存運輸過程中造成的污染[2-6]。隨著農藥使用范圍和使用量的不斷擴大,農藥造成的環境污染和食品藥品安全問題逐漸暴露,如六六六、殺蟲脒、除草醚、艾氏劑、狄氏劑、苯線磷等農藥殘留超標現象時有發生[7]。基于此,該試驗采用GC-MS/MS建立同時測定當歸中35種禁用農藥的殘留檢測方法,以期為控制當歸質量提供數據參考。
1 材料與方法
1.1 試驗材料
1.1.1 ???儀器。7890B-7000D氣相色譜-串聯質譜儀(美國安捷倫公司);ME204/02萬分之一電子天平(瑞士梅特勒-托利多公司);AH40全自動均質器(廈門睿科集團);UPU-1-20T超純水機(四川優普超純科技公司);5810R離心機(德國Eppendorf公司);EVA50A氮吹儀(北京普立泰科儀器有限公司)。
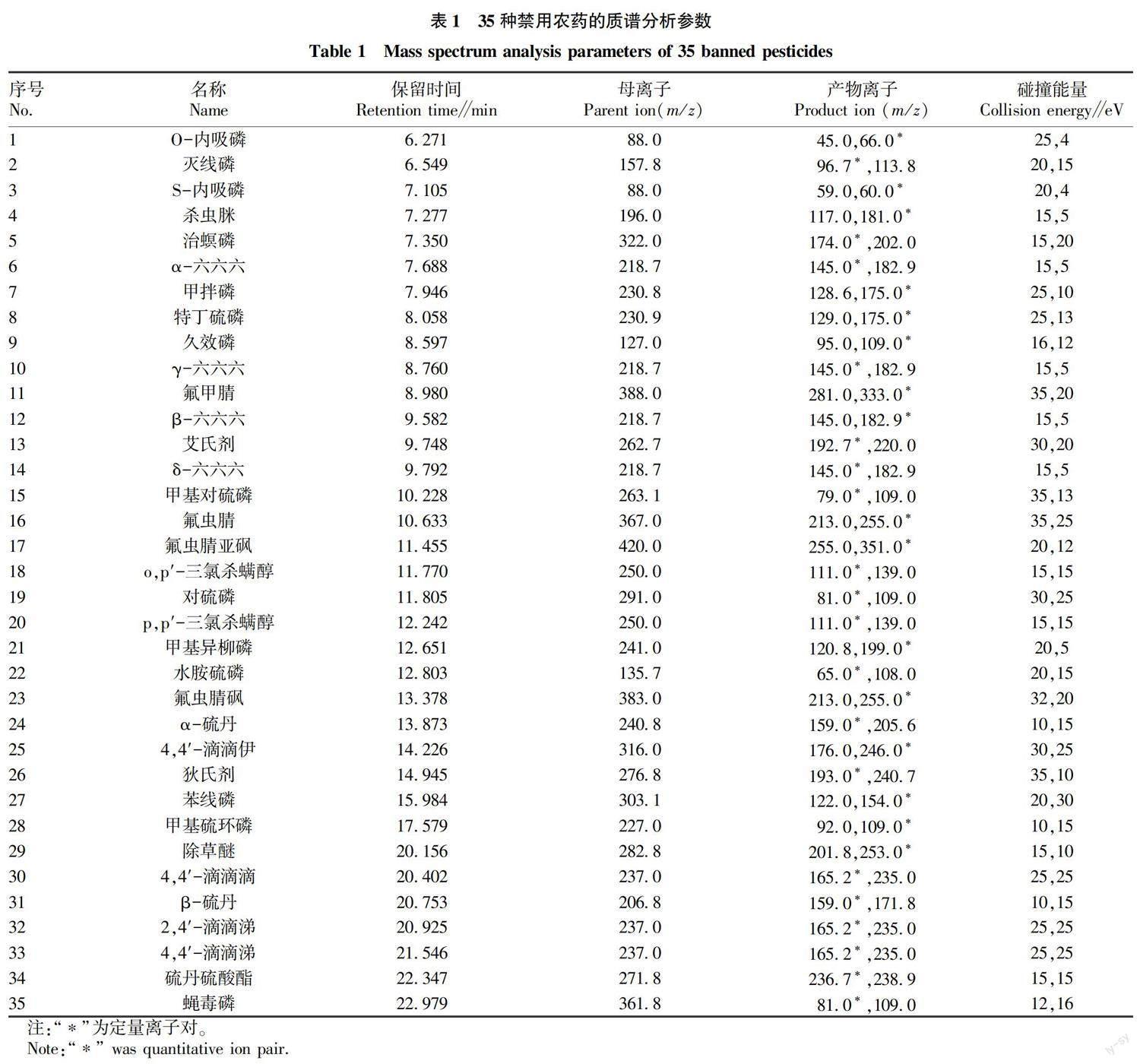
1.1.2 ???試劑。35種農藥對照品:甲基對硫磷(G163528,0.1 g,98.2%)、對硫磷(G130512,0.1 g,98.0%)、久效磷(G833027,0.1 g,98.0%)、α-六六六(G130554,0.1 g,98.1%)、β-六六六(G138918,0.1 g,98.6%)、γ-六六六(G129950,0.25 g,98.8%)、δ-六六六(G132134,0.05 g,99.2%)、4,4′-滴滴涕(G12082000,0.1 g,99.5%)、2,4′-滴滴涕(G12081000,0.1 g,99.6%)、4,4′-滴滴伊(G130552,0.1 g,99.7%)、4,4′-滴滴滴(G123407,0.25 g,98.7%)、殺蟲脒(G150101,0.1 g,99.5%)、除草醚(G149884,0.1 g,98.1%)、艾氏劑(G16913910,100 ng/μL)、狄氏劑(H171083CY,100 ng/μL)、苯線磷(C13421000,0.25 g,99.5%)、蠅毒磷(G92054,0.1 g,98.0%)、治螟磷(G108121,0.1 g,98.9%)、特丁硫磷(G158932,0.1 g,97.8%)、甲拌磷(BCBZ5040,0.1 g,95.4%)、甲基異柳磷(G122317,0.05 g,97.5%)、O-內吸磷(993046CY,10 ng/μL)、S-內吸磷(G1045602,0.1 g,98.3%)、滅線磷(G150818,0.1 g,98.2%)、水胺硫磷(G126442,0.1 g,99.7%)、α-硫丹(G150002,0.1 g,98.1%)、β-硫丹(G139276,0.1 g,99.6%)、硫丹硫酸酯(G1042736,0.1 g,100%)、氟蟲腈(G156586,0.1 g,96.5%)、氟甲腈(13-JQW68-5,0.01 g,98.0%)、氟蟲腈砜(G153662,0.05 g,98.2%)、氟蟲腈亞砜(G141212,0.025 g,99.3%)、o,p′-三氯殺螨醇(219071318,100 ng/μL)、p,p′-三氯殺螨醇(G139873,0.1 g,99.7%),均購自DrEhrenstorfer公司;甲基硫環磷(S033319,100.5 ng/μL),購自Achemtek公司。內標對照品:磷酸三苯酯(G1042718,0.1 g,100%)購自DrEhrenstorfer公司。乙腈為色譜純(德國Merck公司);氯化鈉、冰醋酸為分析純(國藥集團化學試劑有限公司);水為超純水(Milli-Q)。
1.1.3 ???試材。23批當歸(Angelica sinensis Radix)均采自甘肅省定西市岷縣種植地,經蘭州市食品藥品檢驗檢測研究院吳福祥主任藥師鑒定。
1.2 試驗方法
1.2.1 ???混合對照品溶液的配制。分別稱取35種對照品各10 mg,精密稱定,置10 mL容量瓶中,用乙腈稀釋配制成1 mg/mL的對照儲備液,密封儲存于-18 ℃冰箱中。根據各農藥在儀器上的響應靈敏度,準確移取一定體積的對照儲備液,置25 mL容量瓶中,用乙腈稀釋配制成質量濃度為100~250 μg/L的混合對照品溶液,于4 ℃避光保存。精密量取空白基質溶液1.0 mL,平行6份,置氮吹儀上于39 ℃水浴濃縮至約0.4 mL,分別加入混合對照品溶液20、40、100、200、300、400 μL,以乙腈稀釋至1 mL,搖勻即得2~100 μg/L 的6個級別濃度梯度的基質匹配混合對照品溶液[8]。
1.2.2 ???內標溶液的配制。精密稱取磷酸三苯酯內標對照品0.010 44 g,置10 mL容量瓶中,用乙腈稀釋至刻度,搖勻。精密量取上述溶液9.58 μL,置100 mL容量瓶中,用乙腈稀釋至刻度,即得內標溶液。
1.2.3 ???供試品溶液的配制。取供試品粉末(過3號篩)5 g,精密稱定,置50 mL離心管中,加1%冰醋酸水溶液15 mL,陶瓷均質子1顆,渦旋2 min,靜置30 min后,加入乙腈50 mL和氯化鈉1 g,立即搖散,12 000 r/min均質1 min,3 900 r/min離心10 min后取上清液,沉淀重復上述步驟,合并2次上清液,置氮吹儀上于39 ℃水浴氮吹至盡干,用乙腈復溶并稀釋至5 mL,搖勻,過0.22 μm濾膜。
1.2.4 ???色譜條件。安捷倫HP-5MS UI氣相色譜柱(30 m×0.25 mm,0.25 μm);載氣為氦氣,流量1.2 mL/min;程序升溫:初始溫度80 ℃,保持1 min,以40 ℃/min升至200 ℃,再以2 ℃/min升至230 ℃,最后以25 ℃/min升至320 ℃,保持3 min;進樣量1 μL,進樣口溫度280 ℃,不分流進樣。
1.2.5 ???質譜條件。EI離子源,多反應監測(multiple reaction monitoring,MRM)模式,離子源溫度230 ℃,電子能量70 eV,溶劑延遲3.5 min,碰撞氣(氮氣)流速1.5 mL/min。
1.2.6 ???基質效應。對比10、25、50、75 μg/L的當歸空白基質匹配標液與乙腈配制的標液的儀器響應值,以基質標樣的響應值與溶劑標樣響應值的比值計算基質效應。
1.2.7 ???方法學考察。
1.2.7.1 ???線性關系考察。取“ 1.2.1 ”項下當歸空白基質配制的6個級別梯度的混合對照品溶液進行分析,以質量濃度為橫坐標(x)、定量離子對峰面積為縱坐標(y)進行線性回歸計算,繪制標準曲線。
1.2.7.2 ???檢出限和定量限。選取基質匹配標準曲線最低濃度的混合對照品溶液進行稀釋后進樣,以3倍信噪比(S/N≥3)和10倍信噪比(S/N≥10)確定檢出限(LOD)和定量限(LOQ)。
1.2.7.3 ???精密度試驗。選取基質匹配標準曲線最高濃度的混合對照品溶液連續進樣6次,以定量離子對峰面積RSD計算精密度。
1.2.7.4 ???加樣回收率試驗。采用加樣回收法,取經測定未檢出35種農藥的當歸陰性樣品,分3組,精密加入混合對照品溶液適量,考察10、20、100 μg/kg 3個添加水平的加標回收率,每個水平均測定5次,計算RSD。
1.2.8 ???樣品測定。按照“ 1.2.3 ”方法制備23批當歸藥材的供試品溶液,分別精密吸取“ 1.2.1 ”基質匹配混合對照溶液和“ 1.2.3 ”供試品溶液各1 mL,精密加入“ 1.2.2 ”內標溶液0.3 mL,混勻,濾過,取續濾液,注入氣相色譜串聯質譜儀,按照“ 1.2.4 ”色譜條件和“ 1.2.5 ”質譜條件進行測定,每個樣品測定3次,以內標法計算樣品中35種禁用農藥的殘留量。
2 結果與分析
2.1 質譜分析參數
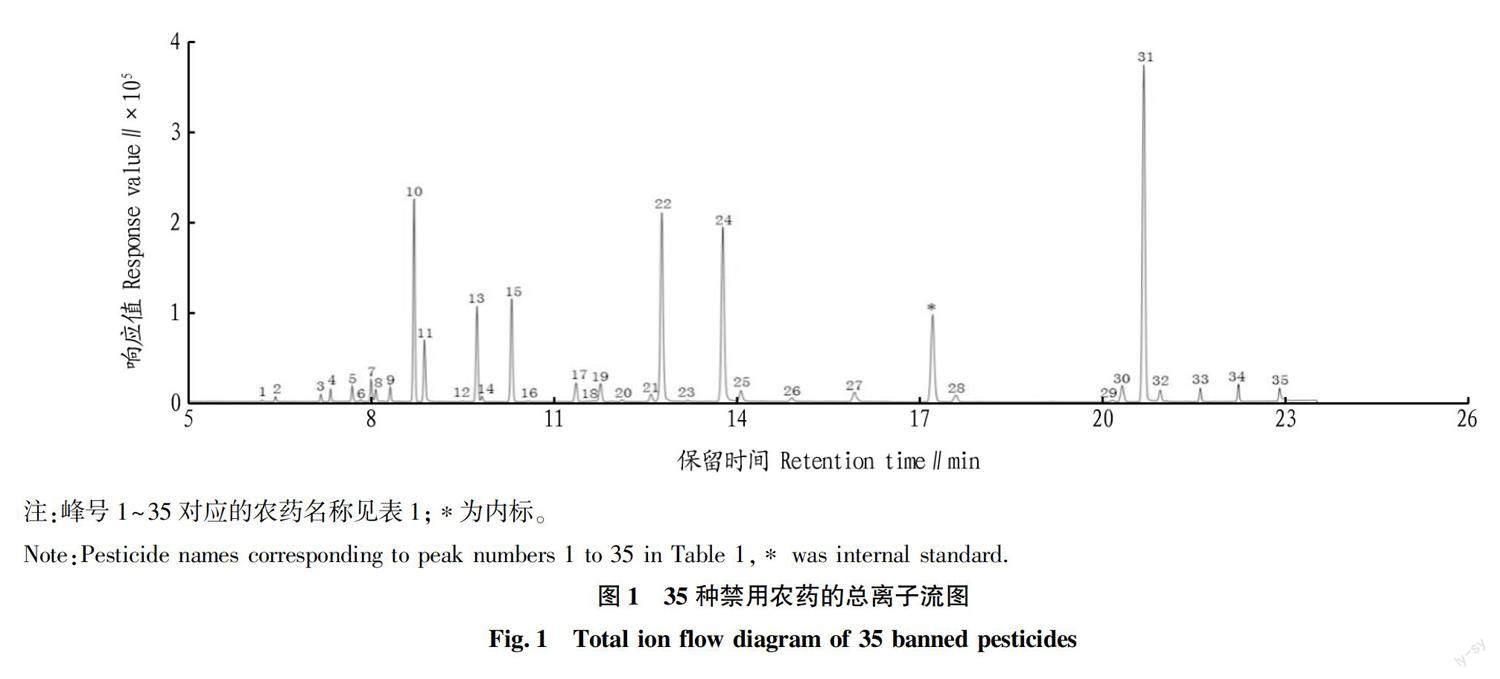
35種禁用農藥的質譜分析參數見表1,總離子流圖見圖1。
2.2 基質效應
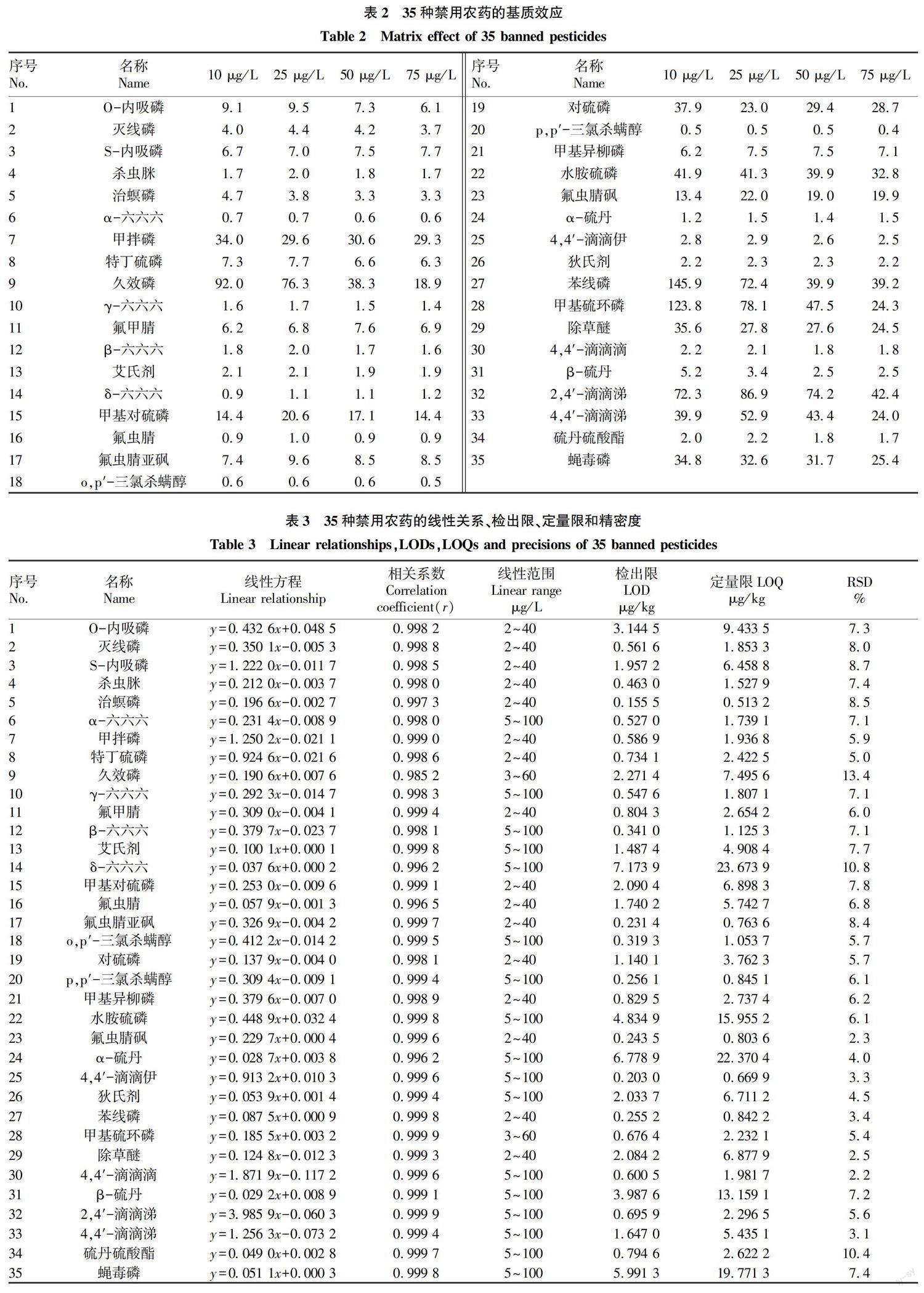
從表2可以看出,α-六六六、o,p′-三氯殺螨醇和p,p′-三氯殺螨醇的基質效應均小于1.0,說明存在基質抑制效應;δ-六六六和氟蟲腈的基質效應接近1.0,說明當歸基質對這2個農藥沒有明顯的抑制或增強效應;其他30種農藥的基質效應均大于1.0,說明存在基質增強效應。因此,為降低基質效應的影響,該試驗采用基質匹配法配制標準溶液并繪制標準曲線[9]。
2.3 方法學考察
2.3.1 ???線性關系。按“ 1.2.7.1 ”方法操作,35種禁用農藥的線性方程見表3,相關系數(r)均不低于0.985 2,表明35種禁用農藥在各自的濃度范圍內呈良好的線性關系。
2.3.2 ???檢出限與定量限。按“ 1.2.7.2 ”方法操作,測得35種禁用農藥的檢出限為0.155 5~7.173 9 μg/kg,定量限為0.513 2~23.673 9 μg/kg,具體結果見表3,表明該方法滿足痕量檢測的要求。
2.3.3 ???精密度。按“ 1.2.7.3 ”方法操作,測得35種禁用農藥定量離子對峰面積的RSD為2.2%~13.4%,具體結果見表3,表明儀器精密度良好。
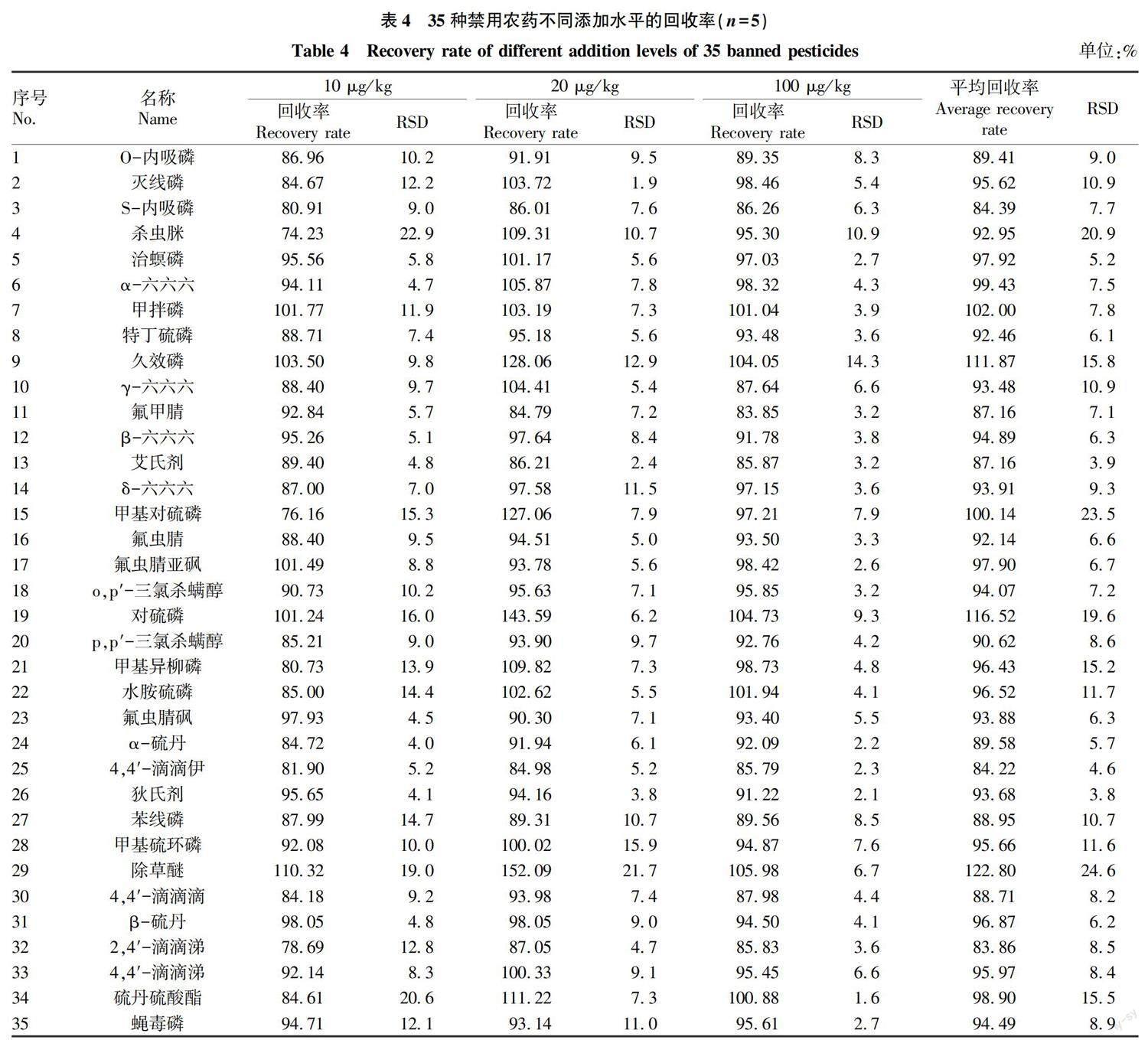
2.3.4 ???加樣回收率。按“ 1.2.7.4 ”方法操作,計算35種禁用農藥的平均加樣回收率為83.86%~122.80%,RSD為3.8%~24.6%,具體結果見表4,表明該方法回收率良好,可用于當歸中35種禁用農藥殘留量的測定。
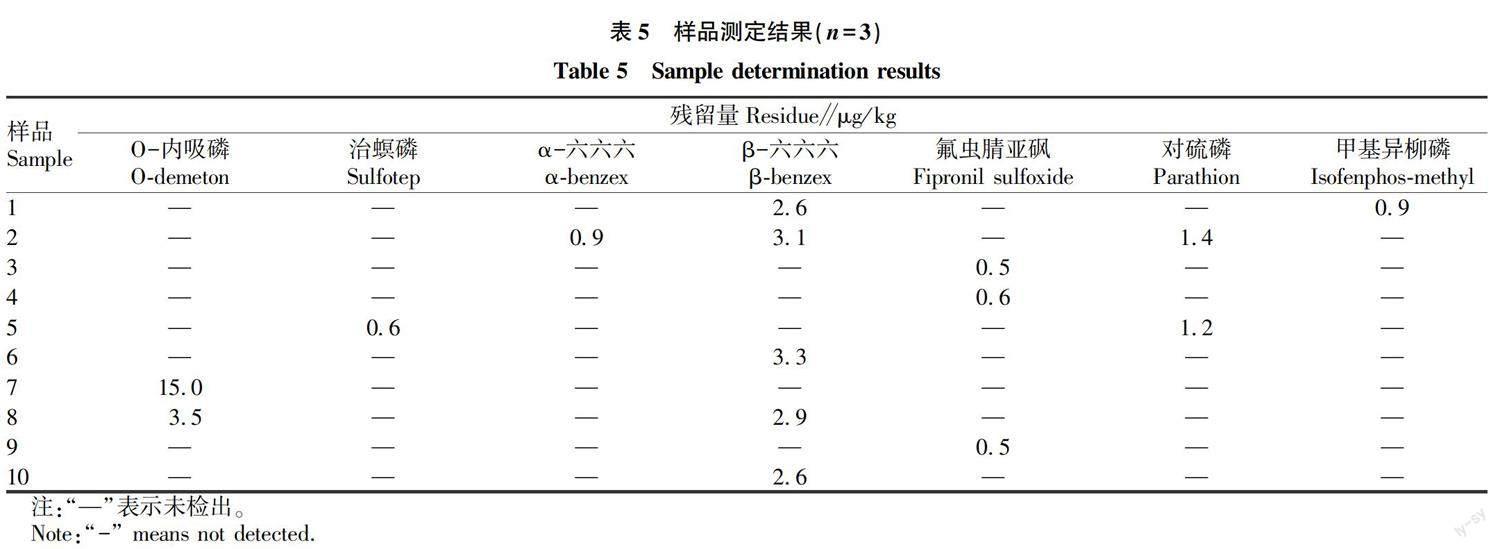
2.4 樣品測定
按“ 1.2.8 ”方法對23批當歸藥材進行測定,結果顯示(表5),有10批藥材檢出O-內吸磷、治螟磷、α-六六六、β-六六六、氟蟲腈亞砜、對硫磷和甲基異柳磷7種禁用農藥。按照《中國藥典》2020版限量標準[10]進行判定,均未超出標準規定。
3 結論
該試驗建立了氣相色譜-串聯質譜同時測定當歸中35種禁用農藥殘留量的檢測方法,具有前處理簡單、靈敏度高、準確快速的優點,同時符合殘留分析的要求,可用于當歸中禁用農藥殘留的定性和定量分析。
參考文獻
[1] ?楊隴軍.甘肅當歸產業發展優勢明顯[EB/OL].(2019-12-23)[2021-07-29].http://www.yidaiyilutcm.org.cn/index.php?m=content&c=index&a=show&catid=52&id=368.
[2] 龐國芳.農藥殘留高通量檢測技術:第1卷 植物源產品[M].北京:科學出版社,2012:530.
[3] 金紅宇,王瑩,孫磊,等.中藥中外源性有害殘留物監控的現狀與建議[J].中國藥事,2009,23(7):639-642.
[4] 李慧君,張文生,吳潔珊,等.中藥材農藥殘留研究現狀[J].中國中藥雜志,2019,44(1):48-52.
[5] 高倩,花日茂,湯鋒,等.中藥材農藥殘留研究現狀[J].安徽農業科學,2008,36(23):10147-10151.
[6] 竇亞潔,劉慧,李曉萌,等.中藥中外源性有害物的殘留現狀及風險評估的研究進展[J].中草藥,2023,54(2):396-407.
[7] 何佩雯,趙海譽,杜鋼,等.氣相色譜技術在中藥農藥殘留檢測中的應用[J].中國實驗方劑學雜志,2010,16(2):126-134.
[8] 陳婷,續艷麗,張文,等.全自動QuEChERS樣品制備系統結合高效液相色譜-串聯質譜法檢測植物源性食品中34種農藥殘留[J].色譜,2019,37(9):1019-1025.
[9] 朱仁愿,劉興國,白雯靜,等.超高效液相色譜-串聯質譜法同時測定黨參中18種植物生長調節劑殘留量[J].分析試驗室,2020,39(12):1450-1455.
[10] ?國家藥典委員會.中華人民共和國藥典:2020年版 四部[S].北京:中國醫藥科技出版社,2020:30-31.
猜你喜歡
中國纖檢(2016年12期)2017-01-20 09:28:19
現代農業科技(2016年20期)2016-12-20 14:51:09
現代農業科技(2016年20期)2016-12-20 09:05:36
分析化學(2016年7期)2016-12-08 00:09:44
分析化學(2016年7期)2016-12-08 00:07:08
價值工程(2016年29期)2016-11-14 01:34:54
科技視界(2016年24期)2016-10-11 18:58:00
考試周刊(2016年39期)2016-06-12 16:01:44
中國科技博覽(2016年4期)2016-04-25 07:25:47
中國科技博覽(2016年8期)2016-04-25 04:57:50
