以生長激素缺乏為臨床表現的先天性無肛伴Kabuki綜合征1例并文獻復習
2023-06-15 07:44:50劉丹如俞曄珩朱芬華
檢驗醫學與臨床 2023年11期
吳 茗,劉丹如,俞曄珩,李 健,朱芬華
復旦大學附屬兒科醫院臨床醫學檢驗中心,上海 201102
先天性無肛屬于先天性肛門直腸畸形(ARM)的一種,大約一半的ARM患者會合并其他器官系統的異常[1-4],但同時合并Kabuki綜合征(KS)和生長激素(GH)缺乏導致身材矮小癥的患兒卻十分罕見。本院收治1例ARM合并KS伴身材矮小的兒童病例,現總結其發病特點、診療思路等,并進行文獻復習,以期提高廣大同行對該類疾病的認識,為臨床診療提供參考依據。
1 臨床資料
患兒,男,5歲10個月,2022年8月10日因發現身材矮小5年余到本院就診。患兒家長自述出生時有宮內窘迫史、腎盂分離和卵圓孔未閉,成長后自愈。出生后診斷為先天性直腸缺如、閉鎖和狹窄(無肛)伴KS。近5年來發現患兒身高低于同齡兒童,生長速度慢,遂就診于本院。擬“身材矮小癥”收入院。就診時查體:患兒特殊面容,眼裂寬,低鼻梁,方顱,高鄂弓,肛門畸形術后改變,身高101 cm,體重16 kg,活動可,智力落后,聽力未通過,戴助聽器。能說句子,不流暢,運動、語言發育稍落后。否認喂養困難史,否認父母近親結婚,無遺傳性疾病史,無矮小家族史(男<160 cm,女<150 cm)。使用Illumina HiSeq平臺進行高通量測序,與人類參考基因組序列對比,結果提示KMT2D基因新發雜合變異。見表1。血、尿、大便常規,肝、腎功能,血脂,電解質,腫瘤標志物,甲狀腺功能檢查均未見異常。胰島素樣生長因子1(IGF-1):44.60 μg/L,IGF結合蛋白3(IGF-BP3):3.30 μg/mL,GH:2.24 μU/mL。超聲檢查提示骨齡約2.5歲。磁共振檢查提示Rathke囊腫,附見胼胝體膝部小囊性灶,雙側側腦室局部稍擴張,胼胝體膝部可疑軟化灶,雙側側腦室擴張。患兒出生后發現肛門閉鎖伴面容特殊,基因檢測提示KMT2D基因新發雜合變異,診斷為KS 1型。患兒今年5歲10月,身高101 cm,低于同齡同性別同地區健康兒童身高的-2SD參考值,身材矮小、勻稱,生長速度減慢,骨齡明顯落后(落后2歲以上),GH激發試驗提示GH缺乏,伴IGF-1減少,診斷為矮小癥。患兒入院后完善各項輔助檢查,空腹行可樂定激發試驗,留取即刻時血液標本送檢GH,予可樂定口服后分別留取30、60、90、120 min時血標本送檢GH,實驗有效,過程順利,評估患兒GH恢復至正常水平后出院。見圖1。
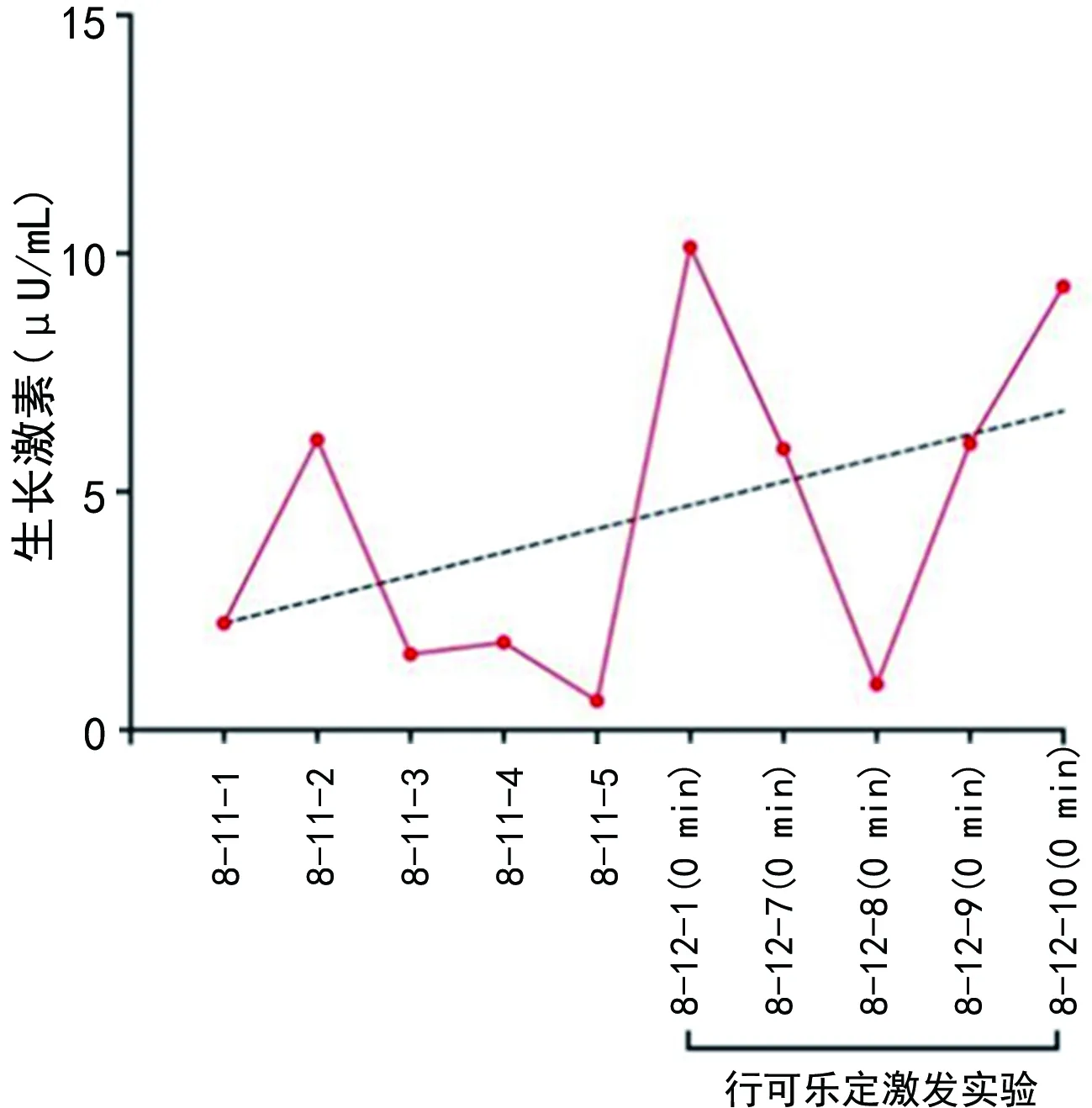
圖1 住院期間血清GH檢測結果變化趨勢
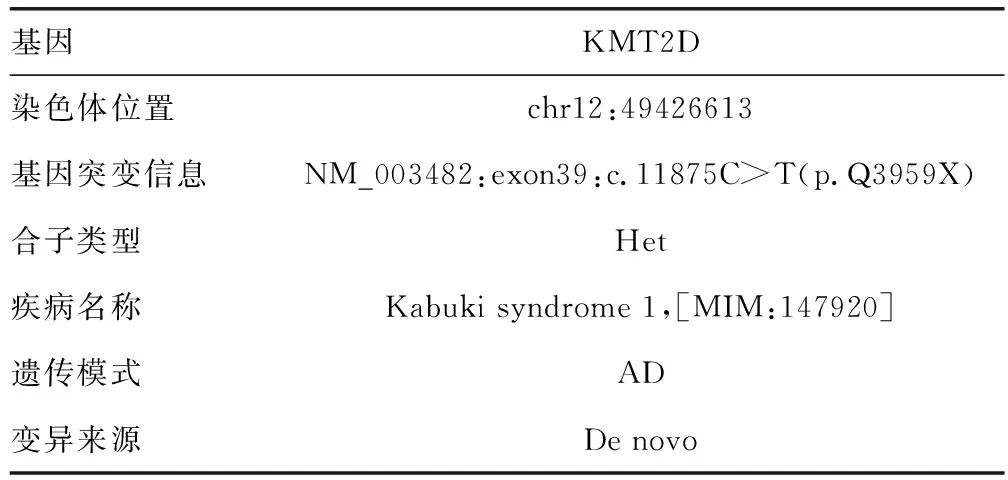
表1 基因檢測結果
2 討 論
大約每5 000個新生兒中會出現1例ARM,且男性多于女性[3],盡管大部分新生兒均會經過產后常規評估,但1/5的新生兒可能出現延遲診斷[2,4]。ARM常合并泌尿生殖系統和肌肉骨骼系統異常[1]。進一步對本例患兒進行基因檢測進行排查。結果顯示,患兒KMT2D基因新發雜合變異,提示KS 1型。KMT2D或KDM6A基因突變是導致KS發生的最主要原因,KS患兒中KMT2D基因突變率高達75%,KDM6A基因的突變占5%的病例(X連鎖顯性遺傳),而約20%病例的病因仍然未知[5-6]。本例患兒經治療出院后又因身材矮小5年余入院就診,完善各項檢查確診為矮小癥。有研究證實,50%~70%的KS病例出生后生長遲緩,有少數病例觀察到產前生長遲緩,絕大多數病例觀察到產后生長遲緩,且與種族無關[7]。此外,有學者指出,KMT2D基因突變的患者身材矮小發生率明顯高于無KMT2D變異體的患者[8]。
以“Kabuki綜合征、KMT2D和矮小”或“Kabuki Syndrome and Short Stature”為關鍵詞檢索中國知網、PubMed等數據庫,檢索時間為2012年10月1日至2022年10月1日,排除基礎研究、綜述、指南單表型研究。檢索到11篇國人發表的文獻[9-19],共24例中國KS同時伴有身材矮小(<-2SD)的患兒,納入包括本例患兒共25例,男∶女=2.1∶1。應用人類表型本體論(HPO)對25例患兒進行臨床表型分型,其中指尖墊(HPO:0001212)16例(64%),反復感染(HPO:0002205)15例(60%),外耳畸形(HPO:0040111)15例(60%),聽力障礙(HPO:0000365)9例(36%),心臟異常(HPO:0002564)9例(36%),脊柱側凸(HPO:0002650)8例(32%),喂食困難(HPO:0011968)8例 (32%),腭裂(HPO:0000175)5例(20%),腎臟異常(HPO:0000834)3例(12%),斜視(HPO:0000486)2例(8%),肛門閉鎖(HPO:0002023)2例 (8%),GH缺乏(HPO:0000824)2例(8%),腺樣體肥大(HPO:0040261)1例(4%),低血糖(HPO:0001943)例1(4%)。文獻復習及本例患兒臨床表征與上述研究結果較為一致,但大部分KS患兒的GH正常,GH缺乏合并各種出生缺陷極為罕見[20]。
本例患兒入院后完善輔助檢查,進行精氨酸、可樂定激發試驗,出院后暫予皮下注射長效生長激素(每周1次)或GH水劑(每晚1次),每3個月定期隨訪內分泌門診,進行健康指導。SCHOTT等[21]發現,經過1年的重組生長激素(rh-GH)治療后KS患者身高有了明顯的線性增長,較早接受rh-GH治療的兒童身高增加幅度更大,KMT2D組與KDM6A組無顯著差異。但長期應用rh-GH治療是否能繼續改善身高、是否可普遍用于矮小且GH缺乏的KS患兒尚需深入研究,尤其是對合并多項出生缺陷基礎疾病的患兒。
綜上所述,本例患兒患有先天性ARM合并KS,同時患有卵圓孔未閉合腎盂分離等多種出生缺陷,生長后又因GH缺乏而矮小,且聽力未通過,極為罕見。在臨床診療中發現ARM或KS時一定要引起高度重視,務必考慮合并其他器官畸形可能,必要時進行詳細檢查,尤其是需要進行基因檢測,對KS患兒應及時檢測其各項代謝和生長指標,并及時進行干預治療。此外對ARM患兒的手術選擇應根據不同疾病特點制訂個體化治療方案。對KS合并矮小癥患兒予以GH治療及生活方式干預,應隨時關注其內分泌代謝異常,通過長期隨訪內分泌門診進行激素水平評估及正確的生活指導,及時發現潛在的風險并阻止其發生,以改善預后,有助于患兒獲得良好的遠期生活質量。
猜你喜歡
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
小讀者(2021年2期)2021-03-29 05:03:48
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
瘋狂英語·新悅讀(2019年11期)2019-12-18 05:14:16
華人時刊(2019年13期)2019-11-17 14:59:54
NBA特刊(2018年21期)2018-11-24 02:48:04
文苑(2018年22期)2018-11-19 02:54:14
